ATTR Amyloidosis
The most common type of systemic amyloidosis is transthyretin amyloidosis (ATTR).
There are two types of ATTR;
- The most common is wild-type (wild type=endogenous/physiologic) ATTR (ATTRwt)
- The rarer ATTR type is inherited ATTR (ATTRv where “v” stands for “variant” gene)
- Approximately 10% of those diagnosed with ATTR in Australia’s AAN clinics have an inherited TTR gene mutation/ATTRv
- There are more than 140 TTR gene mutations that can cause ATTRv
- Cardiac ATTR can be diagnosed without the need for cardiac biopsy in the majority (but not all) cardiac ATTR cases
- There have been recent significant advances in disease modifying therapies for ATTR
- These therapies (Tafamidis, Patisiran, Inotersen) are not currently available through the PBS, but may be accessible via clinical trials or compassionate access programs
Details of the ATTR Amyloid type
Epidemiology
Key points:
- ATTRwt is the most common type of TTR amyloidosis and this is an age-related disorder that is heavily skewed to affect males
- An inherited TTR mutation (ATTRv) is found in ≈ 10% of Australian ATTR patients reviewed in Australia’s AAN clinics
ATTRwt and ATTRv have different epidemiologic profiles.
ATTRwt
- ATTRwt is an age-related disorder
- 25% of those over the age of 85 years in a Finnish autopsy study had ATTRwt as a histopathologic finding1
- Histopathologic identification of amyloid does not translate to symptomatic organ dysfunction until enough of a burden of amyloid is deposited
- The incidence of ATTRwt related symptomatic organ dysfunction is not defined (as yet);
- Epidemiologic knowledge will increase exponentially now that we are entering the era of the “non-biopsy diagnosis” of ATTR
- The median age at diagnosis of cardiac ATTRwt is 79 years 2
- But, ATTRwt can be diagnosed in patients as young as 40 years
- Hence, the historical name, “senile systemic amyloidosis” for ATTRwt is now considered obsolete
- ATTRwt is heavily skewed to occur in males
- males:females was 96:4 In the largest reported ATTRwt cardiac cohort2
- However in other studies the gender gap is much less defined (especially in studies with carpel tunnel syndrome spinal canal stenosis) and this may indicate that it is under diagnosed in females3,4
ATTRv
- ATTRv is a rare autosomal dominant condition that is present all over the world with endemic areas in Sweden, Portugal, Brazil and Japan
- There are >140 TTR gene mutations that can cause ATTR
- Amyloidogenic mutations continue to be discovered at a steady rate
- Regarding incidence of ATTRv in Australia;
- ≈10% of the AAN’s ATTR cohort who undergo genetic testing have an inherited TTR gene mutation
- ≈5% of the AAN cardiac ATTR group have an inherited TTR mutation
- But the true incidence of inherited ATTR is yet to be determined as not every patient with ATTR in Australia undergoes TTR gene mutation testing
- Amyloidogenic TTR mutations are endemic in certain populations but TTR mutations occur “not-infrequently” outside of endemic groups
- The pattern of inheritance and genetic mechanisms for amyloidogenic TTR mutations is the same as for non-TTR inherited amyloidoses;
- Caused by a single base substitution
- Autosomal dominant inheritance pattern
- Incomplete penetrance
- Variable phenotype (even within the same gene mutation)
- Family history is rare (observed in only ≈ 35%) except in endemic populations
- Although amyloidogenic genetic mutations are dominantly inherited the ratio of male to females diagnosed with ATTRv cardiomyopathy has a male preponderance with a male:female ratio of approximately 69:312
- The median age of diagnosis (of non-V122I) ATTRv cardiomyopathy is ≈ 67 years2
- The TTR V122I mutation is endemic is African Caribbean populations and the epidemiology of this group is not relatable to the multi-racial Australian population
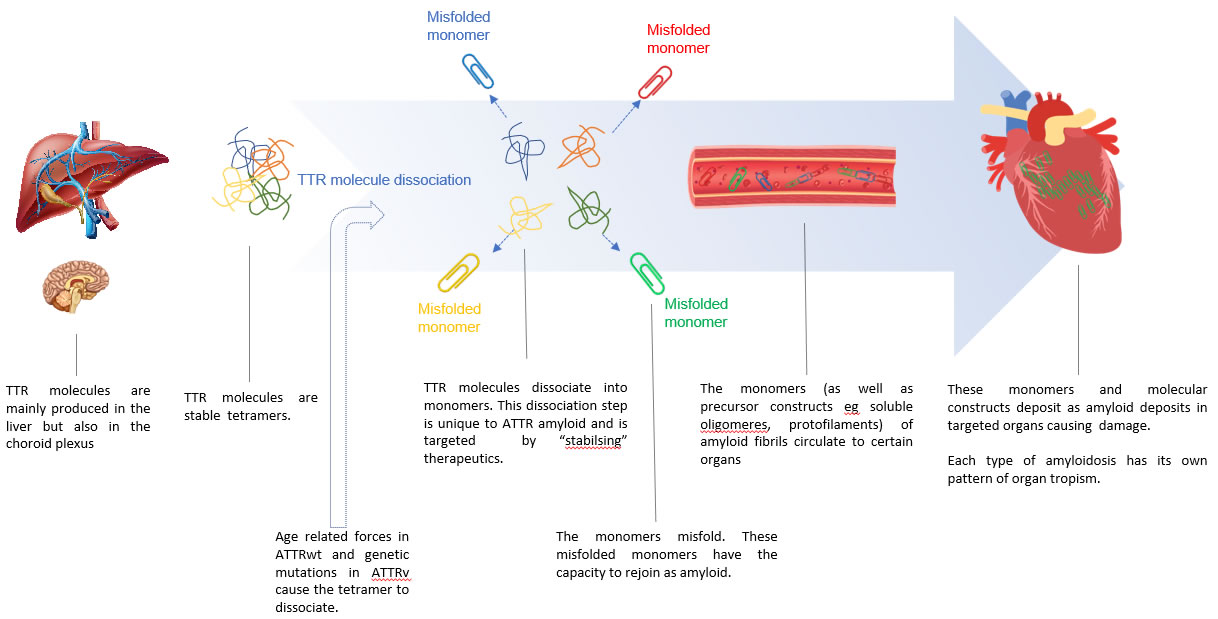
Pathophysiology: The Molecular Process of TTR Amyloid Production
Key Points
- The TTR molecule is a tetramer
- When the TTR tetramer dissociates, its monomers are inherently amyloidogenic with the endogenous ability to form amyloid fibrils
- Transthyretin is a transport protein for thyroxine in the serum and retinol in the CSF (transports thyroxine and retinol)
- The liver produces and secretes TTR in the blood, the retinal pigment epithelial cells secrete TTR into the vitreous of the eye, and the choroid plexus secretes TTR in the CSF
- The liver is the the primary source of TTR
- ATTRwt does not affect the CNS
- CNS and retinal ATTR only occurs with rare amyloidogenic TTR genetic mutations
- The TTR protein is a tetramer
- when the tetramer dissociates into monomers the monomers have the endogenous molecular capacity to misfold and to then form amyloid
- The mechanism that causes dissociation of the TTR tetramer
- In ATTRwt is not understood
- In ATTRv the mutated “variant” TTR tetramer is unstable and dissociates into amyloidogenic monomers
Diagram 1. ATTR formation
Video on ATTR formation
Clinical Presentation and Organ Distribution
Key Points
- The clinical manifestations (and/or the organs affected) differ between ATTRwt and ATTRv
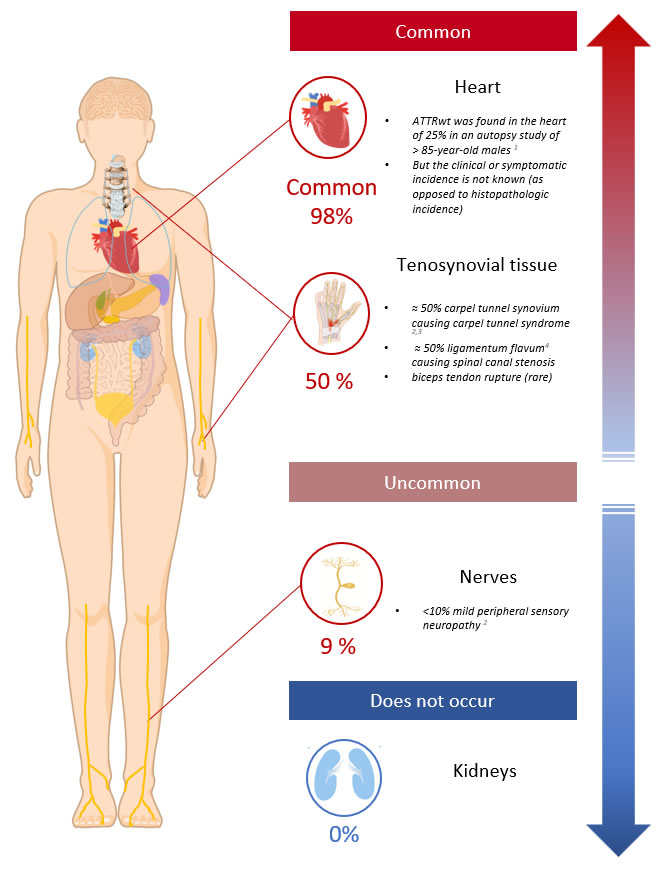
- ATTRwt most commonly deposits in tenosynovial tissues and the heart
- ATTRv most commonly deposits in the heart and nerves
- ATTR (almost) never affects the kidneys, so the presence of proteinuria;
- can help differentiate ATTR from AL
- particularly if patients do not other potential causes for proteinuria for other (common) reasons such as diabetes and hypertension
- Heart ATTR is most commonly ATTRwt
- Nerve ATTR is most commonly ATTRv
- There is a spectrum of clinical manifestations (and/or organ manifestations) in ATTR that differs between:
- ATTRwt vs ATTRv
- The TTR gene mutation involved (in ATTRv)
- ATTR in general, has organ tropism for the heart, tenosynovial tissues and nerves but:
- ATTRwt has particular organ tropism for the heart and tenosynovial tissues
- ATTRv has particular organ tropism for the heart and nerves
- Importantly ATTR almost never affects the kidney (except in rare inherited ATTR mutations) and hence in the absence of concurrent renal disorders the presence of proteinuria can help differentiate ATTR from AL cases
ATTRwt
- The heart Is the most common vital organ affected by ATTRwt
- Neuropathy occurs much less commonly (≈9% of cases) and usually manifests only as a mild peripheral sensory neuropathy6
- ATTRwt has a predilection for the connective tissues and this usually precedes the diagnosis of cardiac involvement by many years;
- Carpal tunnel syndrome is diagnosed in ≈40-50%, on average 10 years before the diagnosis of ATTRwt 6,7
- Spinal canal stenosis can occur from deposition of amyloid in the ligamentum flavum causing spinal cord compression, canal stenosis and nerve root compression
- Rarely rupture of the biceps tendon in the forearm can precede cardiac involvement by many years. This is pathognomonic for ATTRwt.
Diagram 2. ATTRwt Organ Distribution. Showing the clinically significant or symptomatic sites of ATTRwt organ involvement.
Note: Asymptomatic amyloid deposits may be found in the vessels or mucosa at other sites. The ATTRwt organ phenotype is not clearly defined. Knowledge is expected to improve in the ‘non-biopsy diagnosis’ of ATTR era.
ATTRv
- Most ATTRv mutations predominantly affect the heart or nerves
- gastrointestinal symptoms are also common (63%) and mechanisms include both autonomic neuropathy as well as direct gastrointestinal tract tissue involvement. The exact prevalence of the latter is not known.8
- Rarely TTR mutations affect
- CNS (vitreous humour or leptomeninges) as TTR is also produced by the choroid plexus and retinal pigment epithelium
- There are approximately 10 variants of TTR associated with amyloid deposition in the brain and CNS9
- Kidney
- CNS (vitreous humour or leptomeninges) as TTR is also produced by the choroid plexus and retinal pigment epithelium
- The organ sites affected depends on the gene mutation
View Table 1 – Most common TTR gene mutations and organs effected. (Data from the THAOS registry is an international, longitudinal, observational registry that analysed 1579 patients with inherited ATTR/ATTRv.8 - In the past ATTRv was called Familial Amyloid Polyneuropathy or Familial Amyloid Cardiomyopathy but these terms are now obsolete with the recognition that most TTR gene mutations can affect both the heart and nerves to varying degrees
- The TTR gene mutation involved predicts for the nerve vs heart predominance pattern
Diagram 3. Spectrum of nerve and heart involvement in ATTRv. Adapted from10
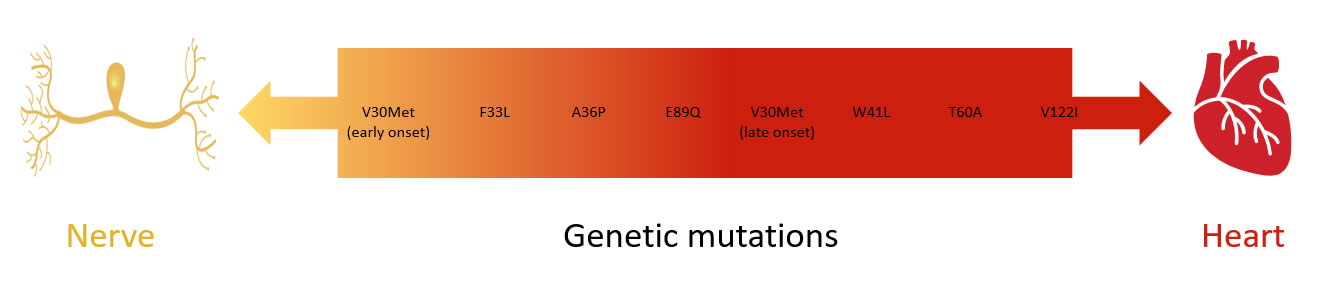
-
- Neurologically, ATTRv often affects all peripheral nerve types (sensory, motor and autonomic nerves) and can be severe and rapidly progressive.
Diagnosis, Staging and Typing ATTR
Key Points
- Amyloidosis can be detected on
- Tissue biopsy
- Cardiac Amyloid Bone Scintigraphy
- Once amyloid has been identified, methods to type the amyloid as ATTR includes;
- Organ screening
- Excluding AL by screening for a monoclonal gammopathy
- Cardiac Amyloid Bone Scintigraphy can type ATTR without the need for tissue biopsy except for the minority with a monoclonal gammopathy
- Anatomical pathology amyloid typing techniques
- Genetic testing is required when ATTR has been identified to differentiate between ATTRwt and ATTRv
- A diagnosis of TTR mutation carriage can be directly made by genetic testing
- This method is employed when screening family members for their family’s known TTR gene mutation
- Genetic screening of those with non-specific idiopathic peripheral neuropathy cannot be currently recommended
- A diagnosis of TTR mutation carriage can be directly made by genetic testing
- Amyloidosis can be detected incidentally on a biopsy of an unrelated organ. The following information pertains to diagnosing ATTR in the patient with suspected cardiac or neurologic amyloid.
- For the basic approach to identifying and then subtyping amyloid please refer to the “What is Amyloidosis>Basic Workup” section of this website
Diagnosing Cardiac Presentation ATTR
- Cardiac ATTR can now be diagnosed without the need for biopsy in the majority of cases using Cardiac Amyloid Bone Scintigraphy
- Cardiologists should consider cardiac amyloidosis in
- Heart failure with preserved ejection fraction (HFpEF)
- Unexplained left ventricular thickening on TTE
- Misleading cardiac ischaemia screening test findings;
- A persistently raised troponin
- A pseudo infarct pattern on ECG (leads V1-V3)
- And especially if the aforementioned are in conjunction with
- Right ventricular thickening
- Refractory atrial fibrillation or CHB, VT/VF, syncope
- Bilateral carpel tunnel syndrome or decompression (ATTRwt)
- Family history of heart failure especially if in association with peripheral or autonomic neuropathy (ATTRv)
- If cardiac uptake is greater than or equal to bone uptake and there is no monoclonal gammopathy then the patient has cardiac ATTR with 100% specificity and 100% positive predictive value 11
- When a monoclonal gammopathy is present the specificity of cardiac uptake on cardiac amyloid bone scan falls to approximately 91%
- This is because some cardiac AL amyloid patients can also exhibit cardiac amyloid bone scintigraphy uptake
- Tests to exclude a monoclonal gammopathy (and all 3 must be ordered) are:
- Serum Protein Electrophoresis + immunofixation (SPEP + IFE)
- Urine Protein Electrophoresis + immunofixation (UPEP + IFE)
- Serum Free Light Chain assay
- Tests to exclude a monoclonal gammopathy (and all 3 must be ordered) are:
- If a monoclonal gammopathy is present a biopsy is required to definitively subtype the amyloidosis
- TTR gene screening is recommended in all identified cases of ATTR Link to Contacts Labs. What is amyloidosis. Genetic screening.
Diagnosing Neurologic Presentation ATTR Amyloidosis
- Neurologists should clinically suspect ATTRv amyloidosis when there is;
- Length dependent sensory-predominant neuropathy with neuropathic pain, especially when in conjunction with
-
- Autonomic involvement
- Small fibre neuropathy
- Unexplained Cardiomyopathy
- Family history of amyloidosis
- Carpel tunnel syndrome/ bilateral carpel tunnel syndrome
- Spinal canal stenosis
- Other : CNS disease is rare (There are approximately 10 variants of TTR associated with amyloid deposition in the brain and CNS).
- Neurologists should consider investigations to evaluate all sizes of nerves, including:
- Nerve conduction studies
- Quantitative sensory testing
- Autonomic studies
- While carpel tunnel syndrome and spinal canal stenosis cause neurological symptoms, these are due to external compression of nerves from tenosynovial infiltration with amyloidosis, hence the presence of these conditions do not indicate an amyloid neuropathy.
- Organ staging is suggested What is Amyloidosis. Organ staging
- If there is cardiac amyloidosis then Cardiac Amyloid Bone Scintigraphy can be used to diagnosis ATTR (often without the need for biopsy)
- If there is no cardiac amyloid then biopsy of involved tissue is required to identify the presence of amyloidosis and to subtype the amyloid using subtyping immunohistochemical stains, +/- mass spectrometry What is Amyloidosis. Basic work-up. Tissue typing
- Screening biopsies (of the rectum, subcutaneous abdominal fat pad, labial salivary gland) has variable sensitivity (reports range between 40-90%) and generally lower sensitivity outside of centres of excellence
- Biopsy of involved sites is generally recommended
- Tenosynovial tissues
- i.e synovoum at the time of carpel tunnel surgery, ligament at the time of spinal canal stenosis surgery
- However carpal tunnel syndrome and spinal canal stenosis can precede symptomatic disease in ATTR, and hence amyloid deposition identified at surgery is not sufficient for the diagnosis of symptomatic ATTR
- Nerves
- Sensitivity of up to 80-93% BUT patchy deposition and can require repeated biopsies at different/directed nerve sites
- Tenosynovial tissues
- When ATTR has been identified by bone scintigraphy or tissue typing Genetic testing is required to differentiate between ATTRwt and ATTRv
- A diagnosis of TTR mutation carriage can be directly made by genetic testing
- This method is employed when screening family members for their family’s known TTR gene mutation
- Genetic screening of those with non-specific idiopathic peripheral neuropathy cannot be currently recommended
Prognosis
- The prognosis depends on:
- The type of ATTR present ie ATTRwt vs ATTRv
- For ATTRwt the prognosis is dependent on the degree of;
- cardiac involvement
- For ATTRv the prognosis is dependent on the;
- degree of heart disease
- degree of nerve disease
- TTR mutation present
- endemic vs sporadic case
- For ATTRwt the prognosis is dependent on the degree of;
- The type of ATTR present ie ATTRwt vs ATTRv
- Prognostic scores are best defined for those with heart disease and applicable to both ATTRwt and ATTRv
Prognosis of Cardiac ATTR
- A prognostic staging system for both cardiac ATTRwt and ATTRv has been developed and validated
- This system utilises the NTproBNP and eGFR 16
- The staging is based on the presence of NTproBNP >3000 ng/L and eGFR <45mL/min
This scoring system stratifies patients between a median OS of 69.2 to 24.1 months Link to Figure 3
The eGFR is thought to decrease in advanced cardiac ATTR due to decreased renal perfusion. NOTE that ATTR does not usually deposit in the kidneys
Additional content not suitable for display on mobile devices available here on desktop or laptopStage NT proBNP (ng/L)* eGFR (mL/min) Median OS (months) I ≥3000 ≤45mL/min 69.2 II All other combinations All other combinations 46.7 III >3000 <45mL/min 24.1 Table 1. Cardiac ATTR amyloid stage and prognosis. Adapted from 16
Prognosis of nerve predominant ATTRv
- The prognosis of those with predominantly ATTRv neurologic disease is less clearly defined
- Untreated cases progress to severe motor, sensory and autonomic impairment, cachexia, imbalance, gait disturbances and limb ulceration
- Hereditary transthyretin amyloidosis is inexorably progressive, with survival of 2 to 15 years after the onset of neuropathy 14,15
- The prognosis depends on:
- the particular TTR gene mutation involved
- endemic (later onset=better prognosis) vs sporadic case (earlier onset=poorer prognosis) 16
Management
Key Points
- Management can be divided into;
- Disease modifying
- Supportive
- For disease modifying therapies;
- TTR stabilisers and TTR synthesis inhibitors have recently been shown to improve organ outcomes
- The future management of ATTR is predicted to consist of a combination of TTR stabilisers and TTR anti-synthesis therapies
Disease Modifying Therapy
Key Points
- Disease modifying therapies for ATTR work via the following mechanisms
- Reducers of TTR synthesis
- TTR tetramer stabilisers
- that stabilise the tetrameric structure of TTR so it cannot dissociate into amyloid forming monomers
- ATTR deposit clearers
Diagram 4. ATTR therapeutic mechanisms.

- ATTR disease modifying therapies are entering a renaissance period;
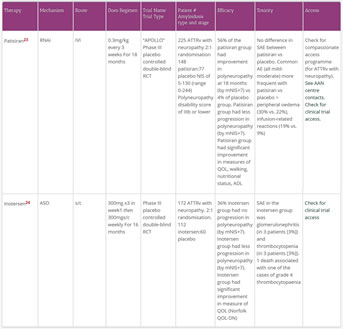
- three RCT trials were published in 2018 which showed the significant efficacy of modern TTR tetramer stabilisers (tafamidis) and reducers of TTR synthesis (patisiran and inotersen)
- The history of genetic therapies has not all been positive. Patisiran and inotersen are genetic therapies that reduce TTR production. A precursor genetic therapy, Revusiran (an RNAi) was associated with increased mortality, in the Endeavour Phase III study
- three RCT trials were published in 2018 which showed the significant efficacy of modern TTR tetramer stabilisers (tafamidis) and reducers of TTR synthesis (patisiran and inotersen)
- Currrently, diflunisal, EGCG and the combination treatment ‘TUDCA and doxycycline’ can be obtained in Australia with varying degrees of administrative and financial requirements. Accessible therapies. Cardiac amyloid for cardiologists>therapy
- Check our Clinical Trials section for access to newer therapies
Reducers of TTR synthesis
- Reducers of TTR synthesis is divided into
- Liver transplantation
- Pharmaceutical reducers
Liver transplantation
- Orthoptic liver transplantation has been used in ATTRv since 1991
- 2202 liver transplants have been recorded on the Familial Amyloidotic Polyneuropathy, World Transplant Registry and Domino Liver Transplant Registry (FAPWTR) up until December 2017
- Of these 26 liver transplants were performed in Australia
- Overall survival (OS) post liver transplantation at 5 years is 77%
- Post transplant, the amount of circulating variant TTR is reduced by more than 95% 17
- Intriguingly (and unfortunately) there is a significantly increased risk of the development of ATTRwt of the myocardium in the transplanted ATTRv patient
- This occurs at a more accelerated rate than in the usual de-novo ATTRwt patient 18
- Val30Met ATTRv patients have better outcomes post liver transplantation with a 10 year overall survival of 74% vs 44% for non-Val30Met ATTRv patients. 19
- Hence, liver transplantation can improve survival in carefully selected patients, but tends to be limited to younger patients (<50 years of age), with shorter disease duration (<7 years), and with the Val30Met variant 20
- In order to preserve the precious resource of donated livers the “domino transplantation” strategy has also been employed
- This is where the ATTRv patients’ livers are donated into a liver failure recipient
- The recipients of the ATTRv patient’s liver are at increased risk of developing ATTR.
- Biopsy-proven de novo amyloidosis occurred in 4/23 donor recipients after a mean of 10 years in one observational study 21
- Liver transplantation rates have been declining over the last few years presumably due to the development of effective TTR synthesis inhibitors (with genetic silencing therapy)
Pharmaceutical reducers of TTR synthesis
- The most effective TTR inhibitors work via gene therapeutic techniques such as RNA interference and antisense oligonucleotide (ASO) genetic therapies
- RNA interference (RNAi) is an endogenous mechanism for controlling gene expression resulting in sequence-specific gene silencing. It is;
- post-transcriptional
- triggered by homologous base pair interaction of double stranded RNA (dsRNA) molecules and target messenger RNA (mRNA)
- multiple types of dsRNA eg microRNA (miRNA) and small interfering RNA(siRNA) can initiate the RNAi pathway
- small interfering RNAs bound to the RNA-induced silencing complex cleave target messenger RNA
- Antisense oligonucleotides (ASO) are short, synthetic, single-stranded oligodeoxynucleotides that can alter RNA and reduce, restore, or modify protein expression through several distinct mechanisms
- RNA interference (RNAi) is an endogenous mechanism for controlling gene expression resulting in sequence-specific gene silencing. It is;
- The development of genetic therapies has been limited by off-target effects including unanticipated changes in downstream signaling cascades and immunologic side effects from viral vectors
- The history of genetic therapies has not all been positive. A precursor genetic therapy, Revusiran (an RNAi) was associated with increased mortality, in the Endeavour Phase III study. 206 ATTRv with cardiac disease were randomised 2:1 to Revusiran: placebo. There were 16 deaths in the revusiran arm and 2 in the placebo. The majority of deaths were cardiac and occurred in older patients with more advanced disease. There was no difference in the baseline characteristics between the two patient groups. The study was closed in 2016.
- In two phase III RCTs published in 2018, the intravenous RNAi patirisan and the subcutaneous ASO inotersen improved neurologic organ function and slowed neurologic deterioration in those with ATTRv 22,23
- A primary endpoint for both of these trials was a measure of neurologic function called the modified Neuropathy Impairment Scale+7 (mNIS+7) which measures sensory, motor and autonomic neuropathy changes
(click thumbnail to view table)
- TTR stabilisers stabilise/maintain the tetrameric structure of the TTR molecule to prevent dissociation into unstable monomers that form ATTR
- The TTR stabilisers assessed in RCTs are tafamidis and diflunisal
- These are both small molecule kinetic stabilisers of the TTR tetramer
- Tafamidis showed a lower all-cause mortality and lower rate of cardiac related hospitalisations vs placebo25
- Diflunisal showed a reduced rate of neurologic progression and improved QOL vs placebo
(click thumbnail to view table)
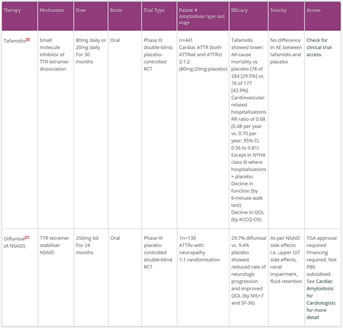
- AG10 is another small molecular kinetic stabiliser that has greater in-vitro activity than tafamidis. It is currently being tested in clinical trials.
ATTR deposit eliminators
- There is limited evidence for ATTR deposit inhibitors
- ATTR deposit inhibitors include:
- combination of doxycycline and tauroursodeoxycholic acid (TUDCA, a bile salt extract)
- doxycycline also has limited evidence for AL deposit elimination
- Epigallaocatechin-3-gallate (EGCG), the active chemical in green tea
- Of note, there are approximately 10 variants of TTR associated with amyloid deposition in the brain and CNS.EGCG presents an attractive treatment option given its human bioavailability of 39%, ability to cross the blood–brain barrier, and excellent tolerability
- combination of doxycycline and tauroursodeoxycholic acid (TUDCA, a bile salt extract)
(click thumbnail to view table)
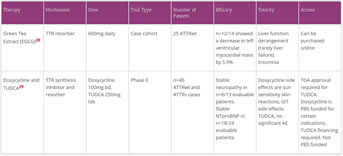
- (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic acid (CPHPC) followed by anti-serum amyloid P component antibody (anti-SAP) will not be discussed;
- This is a pan-amyloid deposit eliminator which has the potential to remove all types of amyloid deposits. Development of this agent is currently stalled.26