The Heart
- ATTR and AL are the main types of heart amyloidosis
- Overall, ATTRwt is the most common type of cardiac amyloidosis
- Note: ATTR is divided into ATTRwt (wild type) and ATTRv (inherited)
- Cardiac ATTRwt, AL and ATTRv have different clinical features, investigation findings and prognosis
- They can be thought of as three different types of cardiac amyloidosis
- Unfortunately, these differing patterns of presentation are not specific enough to differentiate between the cardiac amyloidosis types
- Overall, ATTRwt is the most common type of cardiac amyloidosis
- Cardiac amyloid typing requires a combination of the following essential tests;
- Cardiac amyloid scintigraphy using specific bisphosphonate/bone tracers (for cardiac ATTR)
- Monoclonal gammopathy testing (for AL)
- This is required to screen for the presence of the plasma cell clone that creates AL
- This is also required when performing cardiac amyloid scintigraphy as the presence of a monoclonal gammopathy lowers the specificity of a positive scan result for cardiac ATTR
- as a minority of cardiac AL cases can also show cardiac amyloid scan uptake
- Tissue biopsy and anatomical pathology typing of amyloid deposits
- is required when there is a monoclonal gammopathy to differentiate between
- Cardiac ATTR with an unrelated benign non amyloid forming monoclonal gammopathy
- Cardiac AL caused by a plasma cell clone forming amyloidogenic monoclonal light chain
- is required when there is a monoclonal gammopathy to differentiate between
- A diagnostic algorithm to identify and then subtype cardiac amyloid is presented in the section “Cardiac Amyloidosis for Cardiologists”
- This part of the AAN website will discuss clinical features, cardiac test findings and the management of the most common cardiac amyloidosis types
- Cardiac amyloidosis is often a delayed diagnosis as clinical and test findings can mimic other more common diagnoses
- It is important to diagnose as early as possible as this can result in earlier institution of disease modifying treatment and longer survival
- The most common types of heart amyloid are ATTR (overwhelmingly ATTRwt subtype over the rarer ATTRv) and AL
- ATTR and AL account for over 95% of all cardiac amyloidosis1
- Overall, the most common type of cardiac amyloidosis is ATTRwt
- ATTR is subdivided into
- ATTRwt = wild type transthyretin amyloidosis where the ATTR is made up of physiologic “wild type” transthyretin
- ATTRv = inherited transthyretin amyloidosis where the “v” refers to a “variant” (See ATTR)
- ATTR is subdivided into
- Overall, the most common type of cardiac amyloidosis is ATTRwt
- ATTR and AL account for over 95% of all cardiac amyloidosis1
- Histologically ATTRwt amyloid is found in 10-25% of patients with HFpEF on autopsy studies2 and an even greater proportion of cases are detected in unselected cadaveric case series
- Thus ATTRwt is increasingly referred to as an age-related disorder
- The breakdown of the types of cardiac amyloid referred to AAN clinics is3;
- 63% ATTRwt
- 33% AL
- 3% ATTRv
- 1% “Other” ie AA, non TTR inherited amyloidosis such as apolipoprotein A1
- For ATTR, the most common cardiac amyloidosis, there is now a non-invasive way to diagnose this disorder as well as new disease modifying treatments
- ATTRwt, ATTRv and AL cardiac amyloid are different cardiac diseases4
- There are differences in the pathology, clinical presentation and investigation findings between ATTRwt and ATTRv and AL
- Unfortunately, these differences are not specific enough to subtype the amyloid and cannot be used to replace amyloid typing tests
- The tests that are used to type cardiac amyloidosis are listed below and are required in different combinations;
- Cardiac amyloid scintigraphy using Technetium labelled specific bisphosphonate/bone tracers (for cardiac ATTR)
- Monoclonal gammopathy testing (mandatory for all cases)
- This is required to screen for the presence of the plasma cell clone that creates AL
- This is also required when performing cardiac amyloid scintigraphy as the presence of a monoclonal gammopathy lowers the specificity of a positive scan result for cardiac ATTR
- This is because a minority of cardiac AL cases can also show uptake on cardiac amyloid DPD/PYP scintigraphy
- Hence biopsy and anatomical pathology typing is only required when there is a monoclonal gammopathy in order to differentiate between
- ATTR with an unrelated benign monoclonal gammopathy
- AL with deposits formed from amyloidogenic monoclonal light chain produced by a plasma cell clone
- The management for amyloid cardiomyopathy is distinctly different to that of other cardiomyopathies
- The guidelines for low EF and/or ischaemic cardiomyopathy are not applicable to the infiltrative/deposition disorder of amyloid cardiac amyloid
- Cardioembolic risk is higher in cardiac amyloidosis
- Anticoagulation should always be considered in atrial fibrillation regardless of CHADS-VASc score, and can be considered in cases of severe cardiac involvement without atrial fibrillation
- This part of the website will describe the clinical features, cardiac test findings and the cardiac support of those with cardiac amyloidosis
- A diagnostic algorithm for the identification and typing of cardiac amyloid is discussed in the Cardiac Amyloidosis for Cardiologists section
Amyloidosis and the Heart
Pathophysiology
KEY POINT
- Amyloid deposits in the myocardial extracellular interstitial spaces
- It is an infiltrative disorder with different pathology to cardiac muscle hypertrophic disorders
- Amyloid can eventually deposit in both ventricles and both atria (as well as other areas of the heart)
- The pattern of myocardial deposition differs between ATTR and AL
- Vascular amyloid is more common in AL
- In AL, amyloidogenic light chains are also probably directly cardio-toxic
- Amyloid deposits in the myocardial extracellular interstitial spaces separating and distorting the myocardial cells
- It deposits in the following areas;
- both ventricles
- the presence of right ventricular thickening can differentiate amyloid from processes that usually change the left ventricular parameters alone (such as hypertension)
- both atria
- the valves
- pericardium
- usually leading to small rather than large effusions
- coronary arteries
- classically in the small intramural vessels but not the epicardial coronaries
- both ventricles
- There are differences in the amyloid deposition pattern between the amyloid types
- The myocardial deposition pattern is
- subendocardial and diffuse in AL
- can be patchy and transmural in ATTRwt
- Vascular deposition
- Coronary artery involvement of the small intramural vessels is more common in AL than ATTR5
- The myocardial deposition pattern is
- Myocardial scarring and patchy fibrosis (typical of chronic ischaemic or other non-ischaemic cardiomyopathies) is not described in cardiac amyloidosis
- In cardiac AL disease, research also suggests that circulating AL light chains are directly cardiotoxic6
Clinical presentation
KEY POINT
- Cardiac amyloid deposition may remain symptom free for years (particularly with ATTRwt deposits)
- Cardiac disease usually presents with diastolic heart failure, arrhythmias or conduction abnormalities
- Amyloid related heart failure is usually classified under the heart failure syndrome “Heart Failure with Preserved Ejection Fraction” (HFpEF)
- although reduced ejection fraction can occur in end stage disease
- Few studies have been done in arrhythmias in cardiac amyloidosis
- Atrial Fibrillation is the most common arrhythmia in ATTRwt
- Ventricular arrythmias and sudden death is more common in AL than ATTRwt
- Syncope is also a common presentation but may be non-cardiac in nature and most commonly multifactorial and caused by postural hypotension
- Most commonly cardiac amyloid disease clinically presents with heart failure and/or arrythmias and conduction abnormalities
- A distinction should be made between cardiac amyloid deposition vs disease;
- ATTRwt cardiac deposition is often symptomless for years before causing disease but may still be detected incidentally on TTE or cardiac amyloid bone scan prior to the development of symptoms/disease
- A distinction should be made between cardiac amyloid deposition vs disease;
- The heart failure is typically diastolic in nature
- The heart failure is classified under the heterogenous heart failure syndrome “Heart Failure with Preserved Ejection Fraction” (HFpEF)
- Reduced systolic function is not a common presenting feature but is seen in endstage disease
- The arrythmias can be both bradyarrhythmias and tachyarrhythmias
- Few studies have been done on the electrophysiologic abnormalities and arrhythmias associated with cardiac amyloidosis
- Abnormal electrophysiologic function appears to be most common in the His-Purkinje system
- Progressive conduction system disease is common in ATTR
- Atrial Fibrillation is the most common arrythmia encountered in cardiac amyloidosis. It is present in:
- Ventricular arrythmias are more common in AL10
- Non sustained VT was detected in 27% AL in one holtermonitor study11 but in but only in n=1/20 in an implanted cardiac rhythm recorder study12
- Sustained VT is uncommon
- Sudden death is common in advanced cardiac AL
- The incidence of sudden death appears to be no different between those with or without non-sustained VT11
- Non sustained VT was detected in 27% AL in one holtermonitor study11 but in but only in n=1/20 in an implanted cardiac rhythm recorder study12
- Electromechanical Dissociation (EMD) or pulseless electrical activity (PEA)
- Syncope is common;
- Cardiac causes are;
- Hypotension from the following cardiac contributors
- Reduced stroke volume
- or reduced left ventricular volume from the restricted infiltrated left ventricle)
- Cardiac medications
- over diuresis, anti-hypertensives
- Reduced systolic function
- although this is only seen in end-stage cardiac disease)
- Reduced stroke volume
- Arrythmias: VT, bradycardias, heart block
- Hypotension from the following cardiac contributors
- Other organs involved by amyloidosis can also cause postural hypotension related syncope via
- autonomic dysfunction
- seen in ATTRv and AL (but not in ATTRwt)
- nephrotic syndrome with the osmotic reduction in intravascular volume
- autonomic dysfunction
- Cardiac causes are;
Heart Investigation Findings
- The following heart investigation categories will be discussed:
- Cardiac biomarkers
- Electrocardiogram
- Non-invasive imaging
- Transthoracic echocardiogram
- Cardiac magnetic resonance Imaging
- Cardiac amyloid bone scintigraphy
- Cardiac biopsy
KEY POINT
- Cardiac Biomarkers can provide important clues as to the presence of cardiac amyloidosis
- In advanced cardiac amyloidosis the troponin can become persistently elevated
- The NT-proBNP
- is a more sensitive biomarker for cardiac amyloidosis than the troponin
- and has prognostic value being incorporated into well-validated ATTR and AL prognostic scoring tools
- The cardiac biomarkers NT-proBNP and troponin can become elevated in cardiac amyloidosis
- Both can be spuriously elevated in renal impairment decreasing their utility
- As a rule-of-thumb an NT-proBNP of <1500ng/L is a reasonable negative predictor of cardiac amyloid in those with Stage IV ESRF14
- Both can be spuriously elevated in renal impairment decreasing their utility
- These biomarkers are more sensitive for the presence of AL than ATTR amyloidosis
- But the degree of elevation at presentation (typically higher in AL) is not specific enough to subtype the cardiac amyloidosis
- NT-ProBNP is more sensitive than
- Troponin for the presence of cardiac amyloidosis
- TTE (using standard/basic parameters) for the presence of AL cardiac amyloidosis
- The troponin will remain elevated consistently if caused by cardiac amyloid
- Consider cardiac amyloidosis as a differential diagnosis if Coronary Disease does not explain an increase in troponin
- The NT-proBNP has been well validated for its prognostic value in both ATTR and AL amyloidosis
- The BNP has been validated for its prognostic value in AL14 but not in ATTR
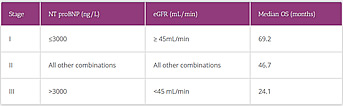
Table 1. ATTR Prognostic Staging Score16
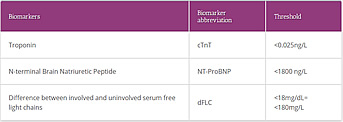
Table 2. Mayo Clinic 2012 AL prognostic parameters17
The cardiac troponin and high-sensitivity cardiac troponin I have been validated for the prognosis of those with AL amyloidosis 17,18
- The following are the most common pathologic findings on ECG;
- Pseudoinfarct pattern
- Low voltage
- Atrial fibrillation
- First degree AV block
- Bundle branch block or intraventricular conduction delay
- The combination of the presence of a low voltage ECG despite increased left ventricular wall thickening on echocardiography is highly suggestive of cardiac amyloidosis (particularly AL cardiac amyloidosis)
- This combination is not so sensitive in ATTRwt as low voltage in limb praecordial leads is less common in ATTRwt cardiac amyloidosis
- The presence of hypertension can decrease the sensitivity of this particular combination by increasing the voltages
- The distribution of ECG findings between the two most common types of cardiac differ (Table 3) but are not specific enough to identify or subtype the cardiac amyloid
| Cardiac ATTRwt % (n=108)* | Cardiac AL % (n=127)20 | ||
|---|---|---|---|
| ECG pathology | % | ECG pathology | % |
| Pseudoinfarct pattern | 63 | Pseudoinfarct pattern | 47 |
| Atrial Fibrillation | 56 | Low voltage in limb/praecordial leads | 46 |
| Low voltage in limb/praecordial leads | 48 | First degree AV block | 21 |
| First AV block | 31 | Atrial fibrillation/flutter | 20 |
| Low voltage in limb/praecordial leads | 22 | LVH | 16** |
| LBBB | 17 | Non-specific intraventricular conduction delay | 16 |
| RBBB | 15 | 2nd or 3rd degree AV block | 3 |
| LVH | 10 | Junctional | 2 |
* Reference 18
Key: ATTRwt= wild type transthyretin amyloidosis
AV= atrioventricular
ECG= Electrocardiogram
LVH= left ventricular hypertrophy
LBBB= left bundle branch block
RBBB= right bundle branch block

- Non-invasive imaging has a central role in the evaluation of patients with cardiac amyloid as its modalities can:
- Signify the presence of cardiac amyloid
- TTE can provide many “alerts” for the presence of cardiac amyloid
- MRI can provide additional more sensitive information for the presence of cardiac amyloid than the basic TTE
- Type the cardiac amyloid
- Cardiac amyloid scintigraphy using certain bisphosphonate/bone tracers can definitively identify and type cardiac ATTR without the need for biopsy in a significant majority of cases;
- This test loses some specificity in those with a monoclonal gammopathy as cardiac amyloid scintigraphy can also be positive in a minority of cardiac AL cases
- Hence, a biopsy containing amyloid and then tissue based amyloid typing tests is required for those with a monoclonal gammopathy
- Cardiac amyloid scintigraphy using certain bisphosphonate/bone tracers can definitively identify and type cardiac ATTR without the need for biopsy in a significant majority of cases;
- Signify the presence of cardiac amyloid
- There are different patterns of non-invasive cardiac test findings between ATTR and AL but these are not specific enough to definitively type the cardiac amyloid
Transthoracic Echocardiogram (TTE)
KEY POINT
- Transthoracic echocardiography provides important and accessible “red flags” for the presence of cardiac amyloid as well as prognostic and response information once the diagnosis has been confirmed
- There are classical TTE findings
- Left ventricular wall thickening without apparent other causes (such as hypertension or valve disease) is an important “red flag” for cardiac amyloidosis
- Other TTE findings can further increase the suspicion for the presence of cardiac amyloidosis
- Left ventricular wall thickening without apparent other causes (such as hypertension or valve disease) is an important “red flag” for cardiac amyloidosis
- Strain assessment provides the more sensitive and specific apical sparing echocardiographic pattern OR “cherry on top” pattern of global longitudinal strain (GLS) in cardiac amyloid
- This type of TTE is not readily available and suspicious classical TTE parameters, in the right clinical settings, can be sufficient to pursue further investigations to confirm the presence of cardiac amyloid
- Echocardiography is usually the main assessment tool that can alert the vigilant cardiologist to the presence of cardiac amyloidosis (Table 2)
- TTE with strain assessment can provide more sensitive and specific indicators of cardiac amyloidosis
- this form of TTE is not readily available everywhere but the strain package is now included in newer echo machines and hopefully this tool will become increasingly more prevalent
| Standard TTE variables | |
|---|---|
| Parameter | Comment |
| Increased LV wall thickness ≥ 12mm | This is a nonspecific finding but in the absence of HT and in the presence of RV wall thickening the specificity increases significantly. The range of “normal” LV wall thickness differs for body habitus and so an IVSd of 11mm may well be pathologic in a petite person. |
| Granular/sparkling appearance of the myocardium | This is neither sensitive or specific as it is a late finding and also seen in other infiltrative diseases |
| Preserved LV ejection fraction > 50% | LV ejection fraction can decrease in end stage disease |
| Right ventricular free wall thickening | This increases the specificity for amyloidosis over causes of cardiac hypertrophy but this feature is limited as; the RV is poorly assessed by TTE; RV hypertrophy can occur from any cause of pulmonary hypertension; can occasionally be seen in HCM (with apical RVH and free wall thickening). In terms of infiltrative disorders, RVH can be seen in Fabry’s disease. |
| Atrial septal thickening | A characteristic finding |
| Normal or small LV cavity | |
| Reduced amplitude A wave | Suggestive of poor atrial function Associated with higher risk of thrombus formation |
| Left and right valve thickening usually causing mild regurgitation | Non specific |
| Small Pericardial effusion | Present in 50% of cardiac amyloidosis cases Non specific |
| Dynamic LV outflow obstruction | A less common feature but it can occur in cardiac amyloidosis and hence dynamic LV outflow obstruction is not only seen in hypertrophic cardiomyopathy |
| Diastology | |
| Parameter | Comment |
| Diastolic dysfunction with mitral inflow pattern that can range from stage 1 (abnormal relaxation) to stage III (restrictive filling pattern) | Diastolic dysfunction is universal |
| Reduced tricuspid annular plane excursion despite normal RV end-diastolic dimension | TAPSE is an early indicator of cardiac amyloidosis in patients with AL |
| High E/A ratio | Severe restrictive mitral restrictive pattern (with E/A ratio > 2) and increased E/E1 is seen later after an abnormal mitral filling pattern |
| Increased left and right atrial volumes and reduced atrial function | A common feature |
| Elevated LV filling pressure | May lead progressively to left atrial enlargement (diameter>23mm/m2, area > 20 cm2 or maximal volume > 28 mL/m2 ) |
| Right atrial enlargement and dilated vena cava reflecting right ventricular (RV) filling pressure | |
| Strain Imaging | |
| Cardiac amyloidosis is associated with reduced global longitudinal strain in the mid and basal ventricle with relative sparing of the apex | This has excellent sensitivity and specificity for cardiac amyloidosis.
The easily recognizable bull’s-eye pattern on polar map can help differentiate CA from other forms of LV thickening such has hypertension or HCM |
| Other | |
| Atrial thrombi | May be seen even in sinus rhythm This occurs more in AL than other types of amyloidosis (51% vs 16%) and is associated with more fatal embolic events (26% vs 8%)23 |
Table 4. TTE features of Cardiac Amyloidosis. Adapted from Falk et al21 and Mohty et al22
Key:
TTE=transthoracic
LV=left ventricle
IVSd=interventricular septal diameter
AL=light chain amyloidosis
TAPSE=tricuspid annular plane excursion
E/EI=the ratio between early mitral inflow velocity and mitral annular early diastolic velocity
- Many of the tabulated TTE features of cardiac amyloidosis are not particularly specific or sensitive
- For the standard TTE the more specific features of cardiac amyloidosis are;
- Right ventricular free wall or atrial septal thickening
- this is rarely seen in left ventricular hypertrophy vs cardiac amyloidosis
- Loss of atrial contraction
- This can be identified by small late diastolic mitral inflow velocity (or small/reduced “A” wave) reduced atrial ejection fraction or reduced atrial strain.
- i.e reduced A wave velocity has been shown to predict the presence of ATTRwt in elderly patients with LVH and sinus rhythm24
- This can be identified by small late diastolic mitral inflow velocity (or small/reduced “A” wave) reduced atrial ejection fraction or reduced atrial strain.
- Right ventricular free wall or atrial septal thickening
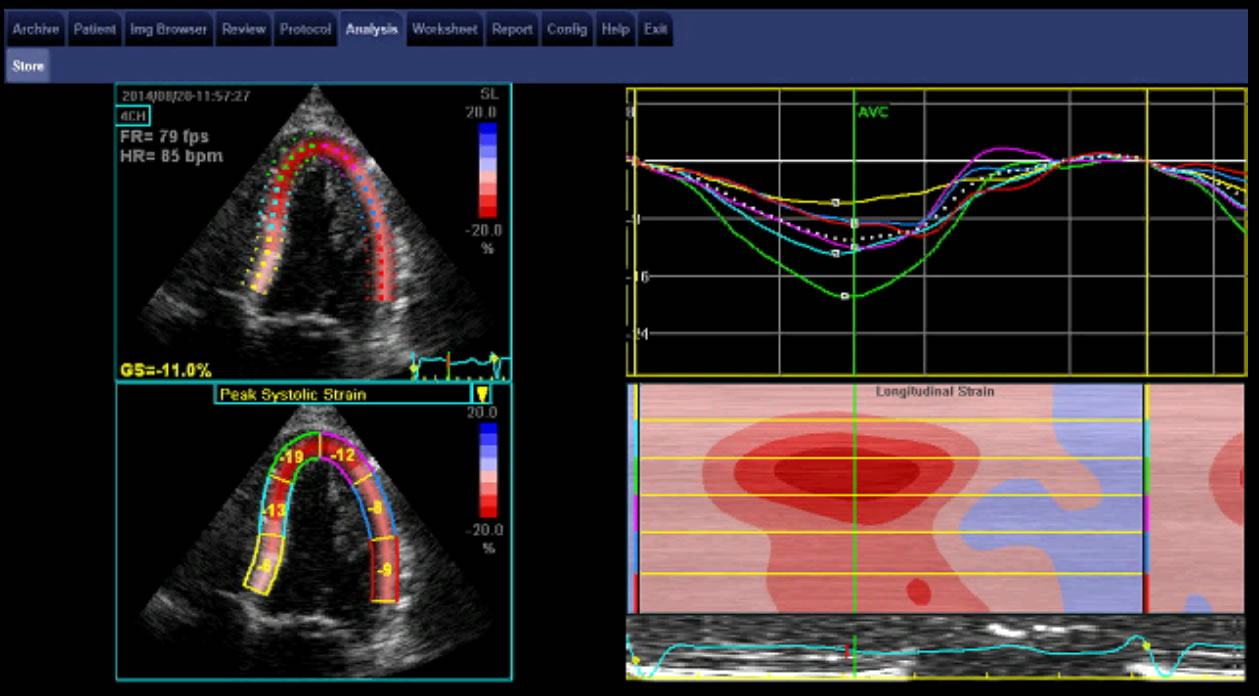
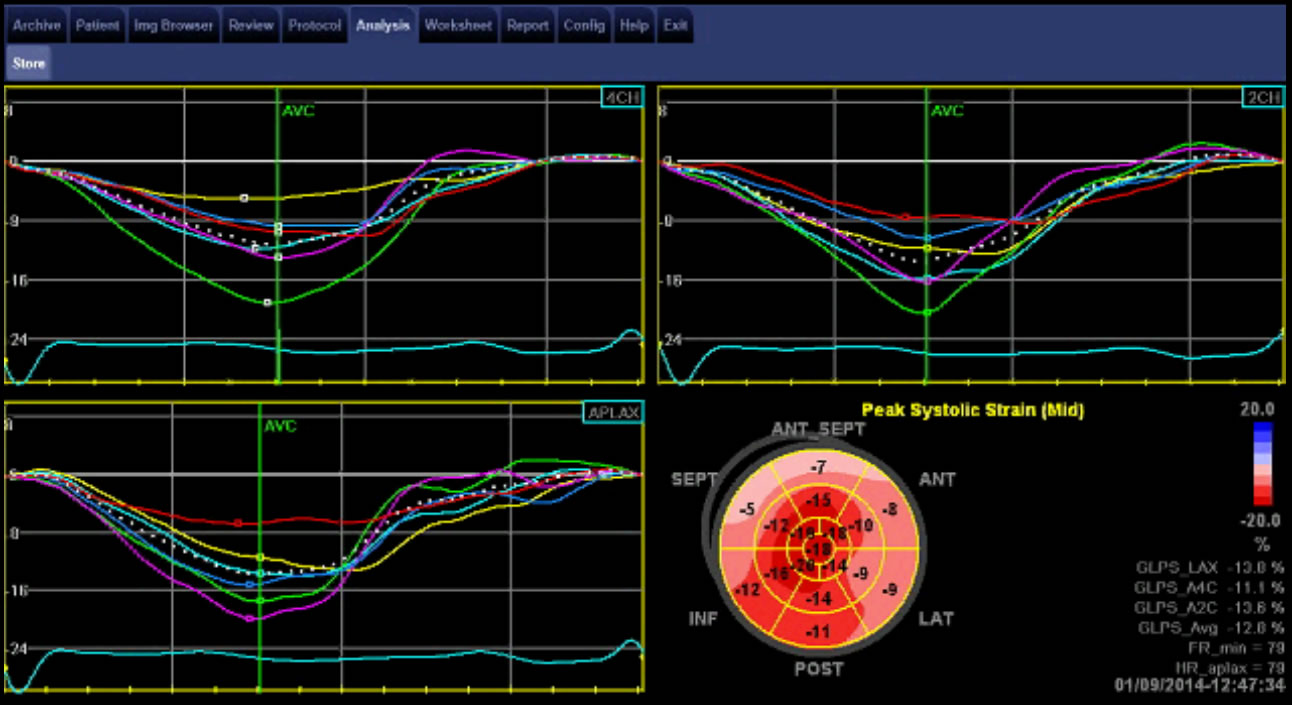
Image 3. The “cherry on top” pattern of reduced longitudinal strain in the mid and basal ventricle with relative sparing of the apex seen in cardiac amyloidosis. Courtesy of Professor Liza Thomas.
- Myocardial strain by 2D speckle tracking echocardiography is recognised as a highly specific and sensitive tool in the evaluation of patients with cardiac amyloidosis;
- Diagnostically a specific and sensitive feature for the presence of cardiac amyloidosis is;
- reduced longitudinal strain in the mid and basal ventricle with relative sparing of the apex, the “cherry on top” pattern of global longitudinal strain (GLS)
- This can distinguish left ventricular infiltration due to amyloid deposition from the ventricular hypertrophy of hypertensive heart disease or hypertrophic cardiomyopathy
- Prognostically, GLS has been shown to be a strong and independent predictor of outcome in patients with cardiac amyloidosis.
- emerging studies show the prognostic value of GLS in addition to the well validated prognostic markers of the cardiac biomarkers for AL cardiac amyloid25
- Diagnostically a specific and sensitive feature for the presence of cardiac amyloidosis is;
- Other relevant points regarding the clinical utility of TTE include:
- That there are differences in patterns of TTE parameters for the different types of cardiac amyloidosis but as per ECG and cardiac MRI patterns of difference, these are not specific enough to replace typing by cardiac amyloid bone scintigraphy +/- biopsy
- TTE can identify atrial dysfunction despite sinus rhythm and this is clinically important as this represents a higher cardioembolic risk where anticoagulation should be considered
- The Interventricular septal diameter (IVSd) is incorporated into the organ staging and response criteria for AL heart disease26
See more in the AL Amyloidosis section
Video: Four chamber Transthoracic Echocardiogram in cardiac amyloidosis. Courtesy Professor Liza Thomas.
KEY POINT
- Cardiac magnetic resonance imaging (MRI) is often performed when cardiac amyloidosis is suspected because of its accuracy with
- spatial resolution allowing accurate morphologic characterisation of volumes and mass
- tissue characterisation through patterns of gadolinium kinetics and T1 mapping
- a characteristic late gadolinium enhancement (LGE) pattern that is diffuse and subendocardial with difficulty in nulling is characteristic for cardiac amyloid
- Cardiac magnetic resonance imaging is more sensitive for identifying features of cardiac amyloidosis when compared to TTE and is very useful where the suspicion exists for cardiac amyloidosis but not enough to proceed with definitive testing
- However, cardiac MRI
- Should not be considered mandatory or absolutely required before proceeding on with definitive tests
- It cannot type the cardiac amyloid
- It is less accessible than TTE
- Due to its accuracy and reproducibility of measurements of LV volumes and mass, cardiac magnetic imaging is superior to TTE and hence is an investigation of choice in the assessment of cardiomyopathies
- Cardiac MRI is also useful for tissue characterisation of the myocardium via Gadolinium kinetics and/or T1 mapping
- Gadolinium kinetics:
- Gadolinium has an interstitial distribution and when cardiac amyloid infiltrates the interstitium this causes gadolinium to stay longer in this expanded space explaining the delayed enhancement (as it is slower to wash out)
- Imaging after administration of gadolinium contrast hence shows a characteristic late gadolinium enhancement (LGE) pattern that is diffuse and subendocardial, and does not follow any particular coronary distribution
- LGE starts in the LV and LA then progresses to the right side of the heart
- The pattern of LGE distribution may also be transmural and patchy
- LGE on CMR is very sensitive for cardiac amyloidosis e.g
- One study identified cardiac involvement by LGE on CMR in 47% of systemic amyloidosis patients with normal wall thickness by TTE27
- Other: Global hyperenhancement on rapid visual T1 assessment, (over 1-2 minutes about 5-10 minutes after gadolinium administration) can be sensitive (93%) and specific (70%) for biopsy proven cardiac amyloid with an overall negative predictive accuracy of 84%27
- T1 mapping:
- Subendocardial T1 is shortened in cardiac amyloidosis.
- Extracellular volume estimation based on T1 mapping can show the extracellular volume expansion of cardiac amyloid (before overt LGE)
- If the myocardium crosses the null point (becomes black) at a T1 timepoint prior to the blood pool, it indicates the presence of diffuse global myocardial hyperenhancement and is characteristic of cardiac amyloidosis
- This creates the technical issue of “difficulty in myocardial nulling”
- Gadolinium kinetics:
- Unfortunately, cardiac magnetic resonance imaging is not as accessible as the TTE
- It is less available
- It requires specialised reporting by both a subspecialised radiologist and imaging cardiologist
- Gadolinium is relatively contraindicated in renal failure (GFR < 30mL/min) due to the 1-2 % risk of systemic sclerosis
- importantly non-contrast T1 -myocardial mapping can be used in this circumstance
- On the other hand, for some patients, it may be more accessible than TTE with strain assessment to confirm suspicions regarding the presence of cardiac amyloidosis
- “Native” (or non-contrast) T1-myocardial mapping techniques indicate the presence of cardiac amyloidosis by showing extracellular volume expansion with significantly increased native T1 times
- This is an important option for those with decreased GFR
- It is less sensitive and specific than gadolinium enhanced images
- Only select cardiac MRI services have the ability and expertise to perform and report T1-myocardial mapping.
- Although features such as ECV expansion and delayed myocardial nulling is quite sensitive and specific to differentiate between cardiac amyloid and cardiac muscle hypertrophic disorders such as HOCM and Fabrys Disease;
- LGE, T1 prolongation and ECV expansion can also be seen in other infiltrative diseases as well as in cardiac inflammation and fibrosis27
- There have been cardiac magnetic resonance imaging studies that have determined algorithms to distinguish ATTR vs AL but there are no validated studies into their positive predictive value29
- Ultimately, as per for ECG and TTE, cardiac amyloid scintigraphy using certain bisphosphonate/bone tracers and/or biopsy are the only definitive methods for the identification and typing of cardiac amyloid
Table 5. Cardiac Magnetic Resonance imaging characteristics of cardiac amyloidosis.
Adapted from Falk et al21
(Click table thumbnail above to enlarge)
Cardiac Amyloid Scintigraphy using Technetium labelled bisphosphonate/bone tracers
KEY POINT
- Cardiac amyloid scintigraphy has a significant diagnostic advantage over the aforementioned imaging modalities as it has the capacity to not only confirm the diagnosis of cardiac amyloidosis but to also specifically type the cardiac amyloidosis as ATTR without the need for biopsy
- Three specific Technetium labelled bone/bisphosphonate/bone tracers have been validated
- DPD, PYP and HMDP
- DPD and PYP are recommended by the AAN
- If cardiac uptake is greater than or equal to bone uptake and there is no monoclonal gammopathy then the patient has cardiac ATTR with 100% specificity and 100% positive predictive value30
- These scans also unfortunately show uptake in a minority of cardiac AL cases (with usually mild uptake seen only)
- Hence in cases with a monoclonal gammopathy, biopsy and tissue typing of the amyloid deposits is required to differentiate between
- ATTR with an unrelated monoclonal gammopathy
- AL with a monoclonal light chain forming the amyloid deposits
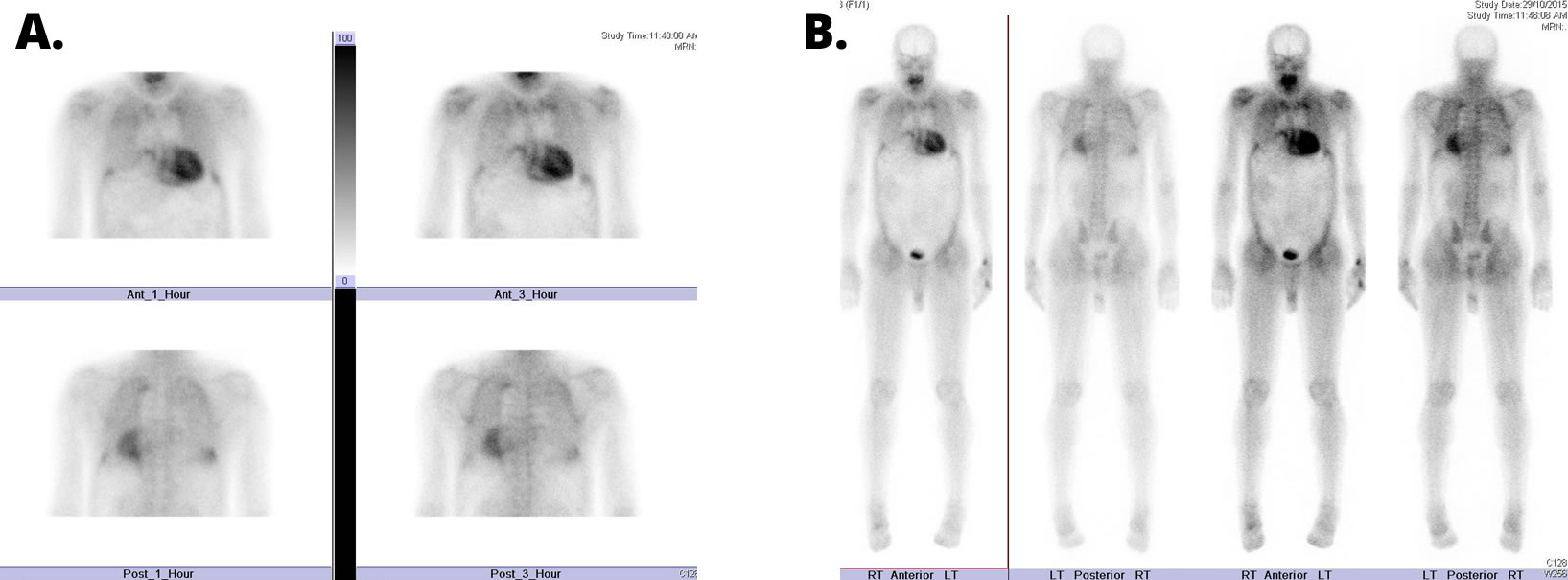
A. 1 and 3 hour spot views
B. 3 hour sweep. Courtesy of Dr David Farlow.
- Cardiac amyloid scintigraphy using Technetium labelled bisphosphonate/bone tracers has been shown to preferentially identify cardiac ATTR amyloid
- the mechanism of uptake seems to be related to the high calcium content in amyloid deposits
- Technetium labelled PYP, a radiotracer used in bone scans, was historically used in cardiology diagnostics to quantify myocardial infarction due to its ability to localize calcium and was referred to as a cardiac “hotspot” scan
- Cardiac amyloid scintigraphy using specific validated Technetium labelled bisphosphonate/bone tracers is now recognised an important sensitive tool that can identify cardiac amyloidosis and also type it highly specifically as ATTR without the need for biopsy
- It has been known from the 1980’s that bone scintigraphy can identify cardiac amyloid31 and subsequent studies showed that certain radiotracers had
- significant uptake in ATTR cardiac amyloidosis
- and no-to-mild uptake in AL cardiac amyloidosis 32
- It is only those with a monoclonal gammopathy who require a biopsy containing amyloid deposits to type the amyloidosis using tissue-based techniques to differentiate between
- ATTR with an unrelated monoclonal gammopathy
- AL with a monoclonal light chain forming AL amyloid deposits
- In 2016, a landmark, large, multicentre study was published that validated the diagnostic utility of cardiac amyloid scintigraphy using bone/bisphosphonate tracers30
- This study showed that, if cardiac uptake is greater than or equal to bone uptake and there is no monoclonal gammopathy then the patient has cardiac ATTR with 100% specificity and 100% positive predictive value
- When a monoclonal gammopathy is present the specificity of cardiac uptake on cardiac amyloid bone scintigraphy falls to approximately 91%
- This is because a minority of cardiac AL amyloid patients can also exhibit cardiac amyloid scintigraphy uptake
- When uptake is present in AL, it is usually milder than in ATTR
- Therefore, it is mandatory to not only perform monoclonal gammopathy testing to screen for AL amyloidosis but in order to interpret the cardiac amyloid scintigraphy result in those provisionally thought to have cardiac ATTR
- This is because a minority of cardiac AL amyloid patients can also exhibit cardiac amyloid scintigraphy uptake
- Cardiac amyloid scintigraphy should use one of the following best validated 99Tc-labelled bisphosphonate/bone markers:
- 3,3 –diphosphosphono-1,2-propanodicarboxylic acid (DPD)
- pyrophosphate (PYP)
- hydroxymethylene diphosphonate (HMDP)
- only twenty one Tc-labelled HMDP patients were included in the cohort studied30
- Hence, the AAN recommends DPD/PYP when the choice exists
- Uptake is graded by
- A semiquantitative visual score of cardiac retention was used in the landmark validation study30 and is also known as the Perugini score
- grade 0 = no cardiac uptake
- grade 1 = mild uptake less than bone
- grade 2 = moderate uptake equal to bone
- grade 3 = high uptake greater than bone
- A quantitative analysis of heart retention was once used in the past but this method has not been well validated for the diagnosis of cardiac ATTR and is not recommended33
- This superseded analysis is calculated by drawing circular regions of interest over the heart, mirrored on the contralateral chest wall and reported as a “heart-to-contralateral ratio”
- A semiquantitative visual score of cardiac retention was used in the landmark validation study30 and is also known as the Perugini score
KEY POINT
- Cardiac biopsy is a sensitive way to detect cardiac amyloidosis
- In the era of the “non-biopsy diagnosis of cardiac ATTR” cardiac biopsy is only required;
- When there is a monoclonal gammopathy of uncertain clinical significance with isolated cardiac disease i.e. when there are no other involved organ sites to biopsy
- For the extremely rare cases of suspected non-ATTR non-AL cardiac amyloidosis where imaging tests (TTE, cardiac MRI) strongly suggest cardiac amyloidosis
- Once there is enough amyloid in the heart to cause ventricular wall thickening, endomyocardial biopsy almost always contains a significant amount of amyloid20
- Endomyocardial biopsy was essentially 100% sensitive for the diagnosis of cardiac amyloidosis in a study that included 33 cases of cardiac amyloidosis34
- However, insufficient biopsy samples or inadequate staining false negatives can occur21
- It is therefore recommended that cardiac biopsy is performed by an operator/ cardiac unit that perform cardiac biopsies regularly and routinely, and interpreted by their pathologists who regularly review cardiac biopsies whenever possible.
- Advanced heart failure / cardiac transplant centres perform these procedures regularly.
- It is therefore recommended that cardiac biopsy is performed by an operator/ cardiac unit that perform cardiac biopsies regularly and routinely, and interpreted by their pathologists who regularly review cardiac biopsies whenever possible.
- The main complication of endomyocardial biopsy is about a 1% risk of right ventricular perforation leading to cardiac tamponade
- Cardiac biopsy is not required to confirm the presence of cardiac amyloidosis when there is:
- Systemic AL amyloidosis with confirmatory biopsy from another organ site and where non-invasive tests (TTE and cardiac biomarkers) are supportive of cardiac amyloidosis
- Positive cardiac amyloid bone scintigraphy in a TTR gene carrier
- Screening by off-target biopsies of tissues such as abdominal fat pad and rectal mucosa has poor sensitivity
Supportive Care of the Heart with Amyloidosis
Introduction
- The management of cardiac amyloidosis is divided into
- Disease modifying therapy, which is specific for each type of amyloidosis and this is further subdivided into
- Anti-synthesis therapies
- directed against the amyloid forming mechanism to reduce further amyloid production, deposition and cardiac failure
- Amyloid clearing therapies
- directed against the amyloid deposit subtype to induce resorption or clearance (these therapies are currently limited). However, if anti-synthesis therapies are successful a proportion of patients may endogenously clear cardiac amyloid TO ADD link to “What is Amyloidosis” section where this concept is discussed
- Disease modifying therapy is directed and targeted against the specific amyloid type and hence differs between ATTR vs AL
- Anti-synthesis therapies
- Supportive therapy, is discussed in this section of the website and mainly addresses
- Heart Failure
- Arrhythmias
- Syncope
- Disease modifying therapy, which is specific for each type of amyloidosis and this is further subdivided into
- Many of the usual generic guidelines for systolic heart failure and arrythmias are not always generalizable to patients with cardiac amyloidosis. This is due to the
- different pathophysiology of amyloid cardiomyopathy i.e. systolic heart failure guidelines for ischaemic heart disease are not applicable to the infiltrative disorder of amyloid
- toxicity/intolerance to many common medical approaches to these conditions
- Negative chronotropic agents are often less well tolerated as cardiac amyloid patients can be dependent on an increased heart rate for cardiac output and are often hypotensive
- There are often other organs affected by amyloidosis further limiting patient tolerance to cardiac therapies i.e
- autonomic neuropathy of ATTRv contributing to hypotension
- nephrotic syndrome of AL contributing to fluid retention
- Cardiac medical therapy is challenging and requires a cautious approach and fine balancing with constant review and adjustment
- As with other causes of diastolic dysfunction, there are no evidence-based supportive therapies that have been found to decrease mortality in cardiac amyloidosis patients, but their quality of life can often be improved significantly with informed and tailored choices in supportive therapies