Amyloid and the Heart
The most critical and fatal manifestation of systemic amyloidosis is cardiac involvement.
Symptoms from cardiac amyloidosis can often be vague leading to late diagnosis and misdiagnosis.
Cardiac Amyloidosis Survey: Patients With AL Amyloidosis and Their Caregivers
Types of systemic amyloidosis in which the heart can be affected:
- AL amyloidosis (second most common organ in AL amyloidosis)
- AA, very rarely seen
- Hereditary amyloidosis below:
- ATTRv
- Apolipoprotein A1
- Wild type ATTR (ATTRwt) amyloidosis
The types of amyloidosis listed above are all very different diseases. But the way the deposition of amyloid affects the functioning of the heart is similar. The progression of heart damage is often slower in ATTRv and ATTRwt than in AL amyloidois.
See separate section on “What is Amyloidosis?”
The normal heart:
- Is a muscular pump about the size of a fist, located in the center left of the chest.
- It is a major part of the cardiovascular system.
What does the heart do?
- Moves blood carrying oxygen and nutrients around the body through a number of interconnecting pipes of various sizes called arteries and veins
- The normal heart expands and contracts about 100,000 times per day pumping five or six quarts of blood each minute, or about 2,000 gallons per day.
- The heart has two sides, left and right, separated by a muscular wall.
The upper chambers are called atria and the larger lower chambers are known as ventricles. - Electrical impulses from the hearts natural pacemaker ignite the heart muscle (the myocardium) and cause the heart to beat (contract).
- This electrical signal begins in the sinoatrial (SA) node, located at the top of the right atrium. The SA node is sometimes referred to as the heart’s “natural pacemaker.”
- When an electrical impulse is released from this natural pacemaker, it causes the atria to contract. The signal then passes through the atrioventricular (AV) node.
- The AV node slows the signal and sends it through the muscle fibers of the ventricles, causing them to contract.
- The right side of the heart pumps blood to the lungs where it receives oxygen.
- The blood then re-enters the left side of the heart from the lungs.
- The heart then pumps the oxygen rich blood around the body.
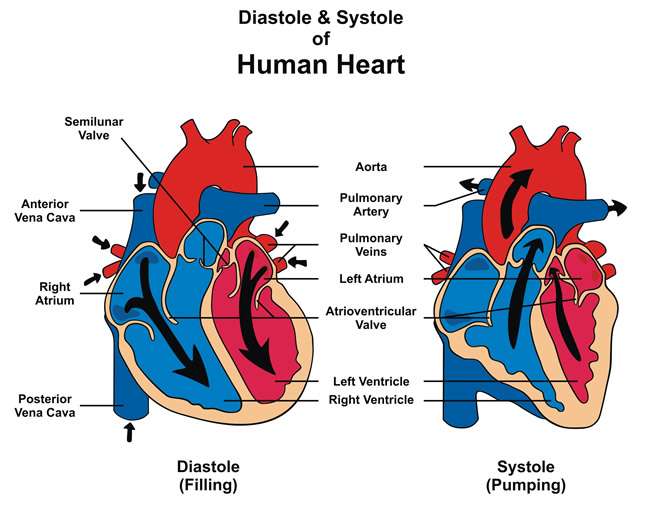
- In healthy people the heart relaxes and contracts between 60 to 100 times a minute.
- When the heart muscles relax the inner chambers fill with blood. When the muscles contract the blood is squeezed out of these chambers into the blood vessels.
What happens when the amyloid protein deposits in the heart?
Cardiac amyloidosis is defined by the presence of extracellular deposition of amyloid with in the heart.
Amyloid deposited in the heart may cause the heart muscle to become stiff over time. If the heart muscle cannot relax enough to fill with blood this leads to accumulation of fluid in the lungs, legs and liver. With time the pumping function decreases, which results in reduced blood flow to the body organs and tissues and may lead to drop in blood pressure.
Heart Failure
- The consequences of cardiac amyloidosis are similar in all types of amyloid. They belong to a group called infiltrative cardiomyopathy and result in progressive heart failure.
- Patients with cardiac amyloidosis typically exhibit heart failure with preserved ejection fraction (otherwise known as diastolic heart failure). This is a common form of heart failure in elderly patients and requires diuretic therapy.
- Cardiac amyloidosis may also affect the way electrical signals move through the heart (conduction system). This can lead to faulty heart signals, (heart block) and may require a pace maker. Abnormal heartbeats (arrhythmias) re also common.
Most types of amyloidosis disease are described as systemic meaning that more than one organ will be effected by the amyloid deposition.
Cardiac amyloidosis therefore is rarely seen alone. Damage in other organs can also impact on the working of the heart.
In AL amyloidosis heart failure may go hand in hand with:
- Peripheral and autonomic neuropathy
- Nephrotic syndrome
- Macroglossia
- Periorbital purpura
In ATTRv heart failure is often seen with peripheral and autonomic neuropathy.
In ATTRwt heart failure is the predominant problem. However, carpal tunnel syndrome, biceps tendon rupture and spinal stenosis may be experienced with atrial fibrillation experienced sometime occurring some time before heart failure.
Symptoms
Symptoms indicating amyloidosis in the heart.
If there is only a small deposition of amyloid in the heart there may be no noticeable symptoms. However, as the deposits build up symptoms will become evident.
These symptoms can often be vague at first and mimic those seen in other diseases which can lead too late diagnosis or a misdiagnosis.
- Fatigue
- Breathlessness on effort
- Swelling of the ankles and feet
- Dizziness
- Weight loss
- Heart palpitations, Atrial fibrillation.
- Black outs
Diagnosis
Treatment
Cardiac Amyloidosis – Treatment Options
Julian Gillmore from the National Amyloidosis Centre in London on Cardiomyopathy in ATTR