The Kidney
The kidney is commonly involved by various subtypes of systemic amyloidoses with light chain amyloidosis (AL) being the most frequent cause of renal amyloidosis followed by amyloidosis A (AA), leucocyte chemotactic factor 2 (ALECT2) then fibrinogen (AFib). (see section on Nomenclature)
However, in certain non-Anglo Saxon ethnic groups the non-AL types of AA and ALECT2 amyloidosis are more common.
There can be many pitfalls in the typing of amyloidosis and the correct typing of renal amyloid is crucial to avoid unnecessary toxicity from misdirected AL chemotherapy.
Each type of amyloidosis is a distinct disorder with different prognoses, renal recurrence risk, extrarenal organ involvement and management.
Proteinuria and renal impairment are the most common manifestations of renal amyloidosis.
A biopsy is required to identify renal amyloidosis. However, in confirmed AL amyloidosis diagnosed by biopsy of other organs, renal amyloidosis can be assumed if there are no other causes for a coincident proteinuria.
Typing the renal amyloidosis requires clinico-pathologic assessment with monoclonal gammopathy testing to screen for AL, organ staging and amyloid protein typing.
In some cases, specialised testing may be required with;
- Amyloid protein typing by mass spectrometry
- Genetic screening for amyloidogenic mutations
Familiarity with the demographic, clinical, renal and histopathological characteristics of renal amyloidosis is required to accurately diagnose and type renal amyloidosis.
The AAN’s clinics and laboratory services were established to assist in the typing and management of amyloidosis and welcome all referrals.
Amyloidosis and the Kidney
Epidemiology
KEY POINT
- Amyloidosis is an infrequent cause of nephrotic syndrome and end-stage kidney disease
- Kidney involvement is common in several types of systemic amyloidosis
- AL is the most common amyloidosis affecting the kidneys causing at least 80% of renal cases, followed by AA, ALECT2 then AFib amyloidosis
Amyloidosis is an infrequent cause of nephrotic syndrome and end-stage kidney disease
Kidney involvement is common in several types of systemic amyloidosis
AL is the most common amyloidosis affecting the kidneys causing at least 80% of renal cases, followed by AA, ALECT2 then AFib amyloidosis
Amyloidosis is an infrequent cause of nephrotic syndrome with an incidence rate that increases with age.1
AL amyloidosis is the most common type of renal amyloidosis accounting for >80% of cases with local data 2 showing a similar distribution of renal amyloid types as observed at the Mayo Clinic 3 (Table 1):
Table 1. Types of Renal Amyloidosis
|
Amyloidosis Type |
AAN (VTAS) Clinic (N=38) |
Mayo Clinic (N=474) |
|
Immunoglobulin derived i.e. AL/AH |
31 (82%) |
407 (85.9%) |
|
Non-AL |
7 (18%) |
67 (14%) |
|
AA ALECT2 AFib |
4 (10%) 2 (5%) 1 (3%) |
33 (7%) 13 (3%) 6 (1%) |
|
Others |
0 (0%) |
15 (3%) |
|
Unclassified AApo Combined* |
|
11 (2.3%) 3 (0.6%) 1 (0.2%) |
Key: AA = Serum amyloid A, AFib = Fibrinogen alpha, AH=Immunoglobulin heavy chain, ALECT2= Leucocyte chemotactic factor-2, AL=Immunoglobulin Light chain, AApo=Apolipoprotein A variants (n=1 for each of AApo AI, II, IV), VTAS=Victorian and Tasmanian Amyloidosis Service
* The one combined case was that of AA amyloidosis and Ig heavy and light chain amyloidosis
However, in certain non-Anglo Saxon ethnic groups AL is not the most common amyloidosis type eg for Egyptian patients both AA (48%) and ALECT2 (31%) are more common than AL (20%). 4
The kidney is commonly involved by systemic amyloidosis but there is a wide range in frequency of renal disease according to the amyloidosis type. The rate of renal amyloidosis in:
- ALECT2 and AFib: is almost universal 4
- AA: is ≈ 97% 5
- AL: is ≈ 70% 6
- ATTR: extremely rare as it is not seen in ATTRwt (the most common type of transthyretin amyloidosis) and is restricted to some ATTRv gene mutations 7
Amyloidosis is an uncommon cause of end-stage kidney disease (ESKD) accounting for approximately 0.8% of patients requiring renal replacement therapy (RRT) in Australia and New Zealand. 8,9 The risk of progression to ESKD is mainly dependent on the amyloidosis type and the availability of disease specific therapy.
Kidney transplantation is rarely performed due the difficulty in ensuring graft survival in the most common type of renal amyloidosis (AL). For instance, less than 1% of ESKD patients with AL amyloidosis received a kidney transplant in the United Kingdom between 1984 and 2009. 10
However, other types of renal amyloidosis are more suitable for the option of renal transplantation as;
- Amyloid recurrence in the renal allograft and/or graft failure may take more than several years to occur eg as in AFib and ALECT2 amyloidosis
- Durable disease modifying therapies may be available eg as in AA
Anatomical Pathology
KEY POINT
- Renal amyloidosis is diagnosed on biopsy using the Congo Red stain followed by examination under polarised light
- Characteristic amyloid distribution patterns in the kidney can provide useful clues to the amyloid type
- Immunohistochemistry (IHC) amyloid typing stains are routinely performed but it is important to note that their positive predictive value is only ≈ 60% and that stains for non-AL/AA renal amyloid types are not readily available
- Laser microdissection of the amyloid deposit followed by mass spectrometry of the constituent protein is used when the amyloid type remains uncertain
Renal amyloid under light microscopy is identified by glomerular deposits of amorphous hyaline material which is Congo Red stain positive displaying apple green birefringence and dichroism effects under polarised light.
In the most common types of renal amyloidosis (AL and AA) the amyloid is deposited predominantly in the glomeruli and vessels and less commonly in the interstitium.
Characteristic patterns of amyloid deposition can be useful in typing other renal types of amyloid however these features can overlap and are not pathognomonic. The characteristic glomerular deposition patterns can be summarised as follows:
- AFib: Vascular
- ALECT2: Cortical interstitial
- AApo: Medullary interstitial
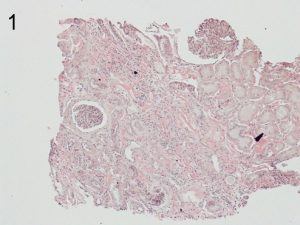
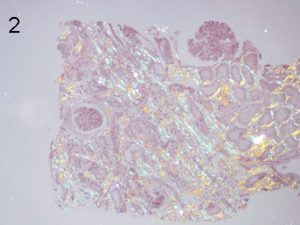
Photos 1 and 2. ALECT2. The cortical interstitial deposition pattern of Congo Red staining (1) of ALect2 renal
amyloid under light microscopy and polarized light (2). Photos courtesy of Dr Shannon Bahamehr Fadaee.
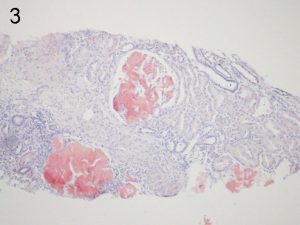
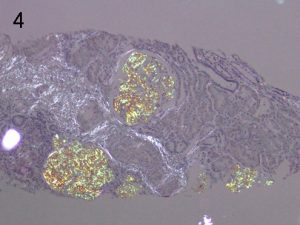
Photos 3 and 4. AFib. The glomerular deposition pattern of Congo Red staining (3) of AFib renal
amyloid under light microscopy and polarized light (4). Photos courtesy of Dr Shannon Bahamehr Fadaee.
On electron microscopy the amyloid is characterised by randomly arranged, small organised fibrils, with a diameter of 8-15 nm. The alternative diagnosis of fibrillary glomerulonephritis can be differentiated by;
- A lack of positive Congo Red staining
- Larger fibrils (12-24 nm) in diameter although there is an overlap
- Positive DNAJB9 immunohistochemistry (IHC) staining which has 98% sensitivity and >99% specificity for fibrillary glomerulonephritis 11
IHC amyloid typing stains should always be performed. However, it is important to acknowledge that false positives and false negatives occur due to intrinsic limitations in the antisera used and that the positive predictive value of these stains is only in the order of ≈ 60%. 12,13 See anatomical pathology section The basic IHC typing panel for renal amyloid includes antisera against the following amyloid fibril proteins:
- Lambda and Kappa light chain
- AA
Immunohistochemical amyloid typing stains for rarer amyloid proteins are not readily available in Australia eg the ALECT2 and AFIb stains. And non-AL nan-AA cases are typically referred to reference labs for further typing by second tier IHC stains and if required mass spectrometry. See special tests section
Clinical Presentation
KEY POINT
- Renal amyloidosis usually presents with proteinuria and/or impaired kidney function
- The amyloid type and amyloid burden and its distribution in the kidney influence the clinical presentation
- The most common type of renal amyloidosis is AL amyloidosis and this typically presents with multiorgan disease
- The next most common types AA, ALECT2 and AFib are usually associated with isolated kidney disease
The usual presenting features of renal amyloidosis are proteinuria and/or impaired kidney function. However, each type of renal amyloidosis can be considered as separate disorders with different levels of proteinuria, rates of progression to ESKD, extra-renal manifestations and therapy.
The proteinuria may be mild through to nephrotic in range. ALECT 2 often presents with minimal proteinuria (as the amyloid is predominantly interstitial) and typically remains subnephrotic even when there is progression to ESKD. The proteinuria of AL and AA is of a glomerular pattern with the capacity to develop nephrotic range proteinuria.
The kidney dysfunction can range from minimal to end-stage kidney disease (ESKD) requiring dialysis. The rate of progression to ESKD also varies and is slower in ALECT2 than in AL with ALECT2 associated with a mean eGFR loss of only 4.2 mL/min/yr. 15
AL is commonly associated with multi-organ amyloidosis whereas the other types of are more likely to present with isolated renal disease. Of important prognostic significance, the heart is commonly involved in AL but only rarely in AA (≈2%) and is not described as yet in ALECT2.
There are other clinical features that can assist in the typing of the renal amyloid;
- ALECT 2 is typically seen in non-Anglo Saxon ethnic groups, particularly patients of Egyptian, Hispanic, Indian and Pakistani background (and it has also been recently described in the Chinese population) 16
- AA amyloidosis is classically associated with disorders causing uncontrolled inflammation eg an active autoinflammatory disorder such as rheumatoid arthritis, chronic infection or inflammatory malignancies (typically renal cell cancer.) However, it has become increasingly common for the inflammation to be of uncertain origin, a phenomenon which may be partly related to obesity and its associated low-grade chronic inflammation.
For hereditary amyloidosis there may not be any clear family history eg there is no family history in >50% AFib despite an autosomal dominant inheritance pattern and this is partly due to the incomplete penetrance pattern of the hereditary amyloidoses. 17
The Workup and Typing of Renal Amyloidosis
KEY POINT
- After a kidney biopsy has identified amyloidosis the basic workup starts with:
- Screening for AL with monoclonal gammopathy testing
- and
- Organ staging
- Renal involvement may confound the interpretation of some of the basic workup tests
- Monoclonal gammopathy screening is an essential part of the assessment of renal amyloidosis, but caution is required with interpretation of the serum free light chains (SFLC) in the setting of impaired kidney function
After a kidney biopsy has identified amyloidosis the basic workup starts with;
- Screening for AL by assessing for a monoclonal gammopathy
- and
- Organ staging
The components of the basic workup (Diagram 1) are discussed in more detail elsewhere.
Diagram 1. The Basic Workup after Identification of Renal Amyloidosis
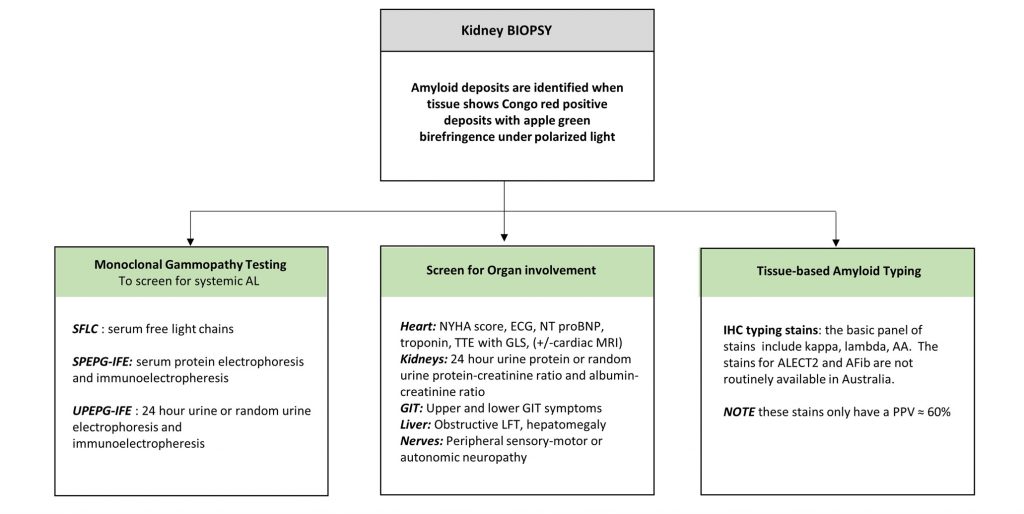
Key: AA- = Amyloid A, AFib = Fibrinogen amyloid, amyloid ALECT2 = Leucocyte chemotactic factor 2 amyloid, ECG = electrocardiogram, GIT= gastrointestinal tract, GLS= global longitudinal strain, LFT=liver function tests, MRI= magnetic resonance imaging, NT proBNP= N-terminal pro brain natriuretic peptide, PPV= positive predictive value, TTE= transthoracic echocardiogram
A summary of the different diagnostic findings for AL vs non-AL renal amyloidosis is diagrammatically presented (Diagram 2).
Diagram 2. The Test findings of AL vs non-AL Amyloidosis
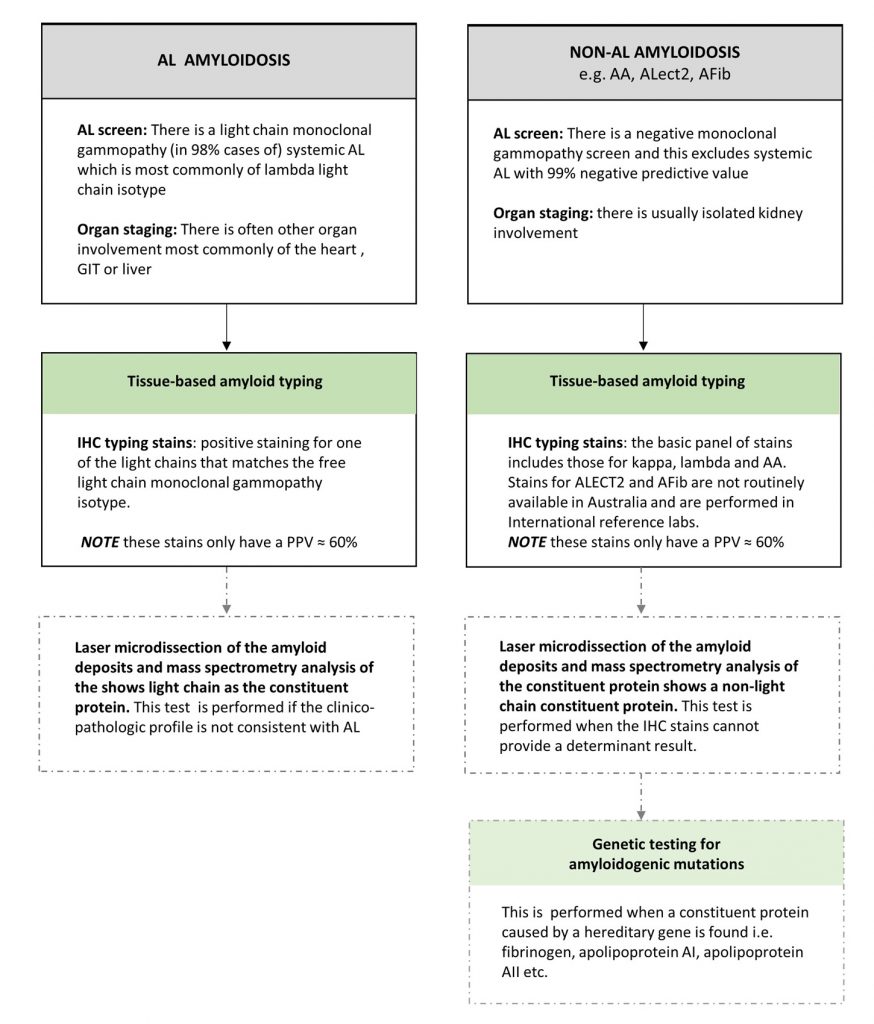
![]()
AA= Amyloid A, ATTR= transthyretin amyloid, ECG = electrocardiogram, GIT= gastrointestinal tract , GLS= global longitudinal strain, IHC= immunohistochemical, Ix= investigation, LFT=liver function tests, MGUS= monoclonal gammopathy of uncertain significance, MRI= magnetic resonance imaging, NT proBNP= N-terminal pro brain natriuretic peptide, PPV= positive predictive value, TTE= transthoracic echocardiogram
Renal failure can confound the interpretation of many of the basic workup tests and the issues are discussed below.
Clinicopathologic profiles
There are helpful clinico-pathologic characteristics for the different types of renal amyloid but they should not be used to definitively type the amyloidosis (Table 2).
Table 2: Clinical and histopathological characteristics of the most common types of renal amyloidosis
| Type | Renal and clinical presentation | Renal compartment involved | Supporting/ confirmatory tests |
| AL | Proteinuria, nephrotic syndrome, impaired kidney function
+/- cardiac amyloid |
Glomerular + Vascular > Interstitium | Light chain monoclonal gammopathy (usually lambda)
Light chain (usually lambda) restriction on IHC staining. Note: Light chain IHC stains only have a PPV of approximately 60% |
| AA | Proteinuria, nephrotic syndrome, impaired kidney function
Inflammatory conditions i.e. rheumatoid arthritis, chronic infection, renal cell cancer, obesity |
Glomerular + Vascular > Interstitium | AA on IHC staining
Raised Serum Amyloid A and/or CRP |
| ALECT2 | Often minimal proteinuria, impaired kidney function
Median age at presentation of 62-68 years Specific for certain non-Anglo Saxon ethnic groups (Egyptians, Hispanics, Indians and Pakistani) |
Interstitium +++ | IHC staining for LECT2 is reliable but not available in Australia
Mass spectrometry showing LECT 2 protein as the constituent amyloid protein is often required |
| AFib | Proteinuria, impaired kidney function
Median age at presentation of 58 years No family history in >50% |
Glomerular +++ | IHC staining for Fibrinogen (not easily available in Australia)
Mass spectrometry showing fibrinogen protein as the constituent amyloid protein is often required Genetic testing of the Fibrinogen exon of interest |
Key; AA= serum Amyloid A amyloid, AL= light chain amyloid, ALECT 2= leucocyte chemotactic factor amyloid, CRP= C reactive protein, IHC= immunohistochemistry, PPV= positive predictive value
Prognosis
KEY POINT
- The patient and renal prognosis varies according to the amyloidosis type
- Amyloid specific therapy is available for AL and AA
- Early treatment is crucial as deep suppression of amyloid synthesis is associated with organ responses and improved renal and patient survival outcomes
- Renal amyloid clearance can occur after disease modifying treatment, but achieving a renal response often takes more than 6 months after stopping the amyloid forming process
The patient’s overall survival and renal prognosis varies according to the;
- Type of amyloidosis
- Degree of renal involvement
- Availability and efficacy of therapy
The renal prognosis can improve with treatment as renal amyloid clearance can occur by the proteolytic breakdown of amyloid fibrils by phagocytic cells if amyloid production is stopped (or markedly reduced) by disease modifying treatment. This process is slow and achieving a renal response often takes more than 6 months after an effective reduction in amyloid synthesis. Disease modifying therapy and hence the chance of renal recovery exists for both AL and AA amyloidosis.
Supportive Care
The management of renal amyloidosis can be divided into disease modifying treatment and supportive care. Disease modifying therapy is described elsewhere on this website under each type of amyloidosis.
The supportive care of nephrotic syndrome is discussed in this section of the website and addresses;
- Fluid retention
- Thromboembolism
- Hyperlipidaemia
Generic supportive therapies for nephrotic syndrome are not always generalisable to patients with renal amyloidosis.
Renal replacement therapy is discussed and divided into;
- Dialysis
- Kidney transplantation
Palliative care (without dialysis) should be considered for those with poor prognosis and quality of life.
Renal supportive care is challenging and requires a cautious approach with constant review and adjustment.