The Basic Workup and Approach
General Overview
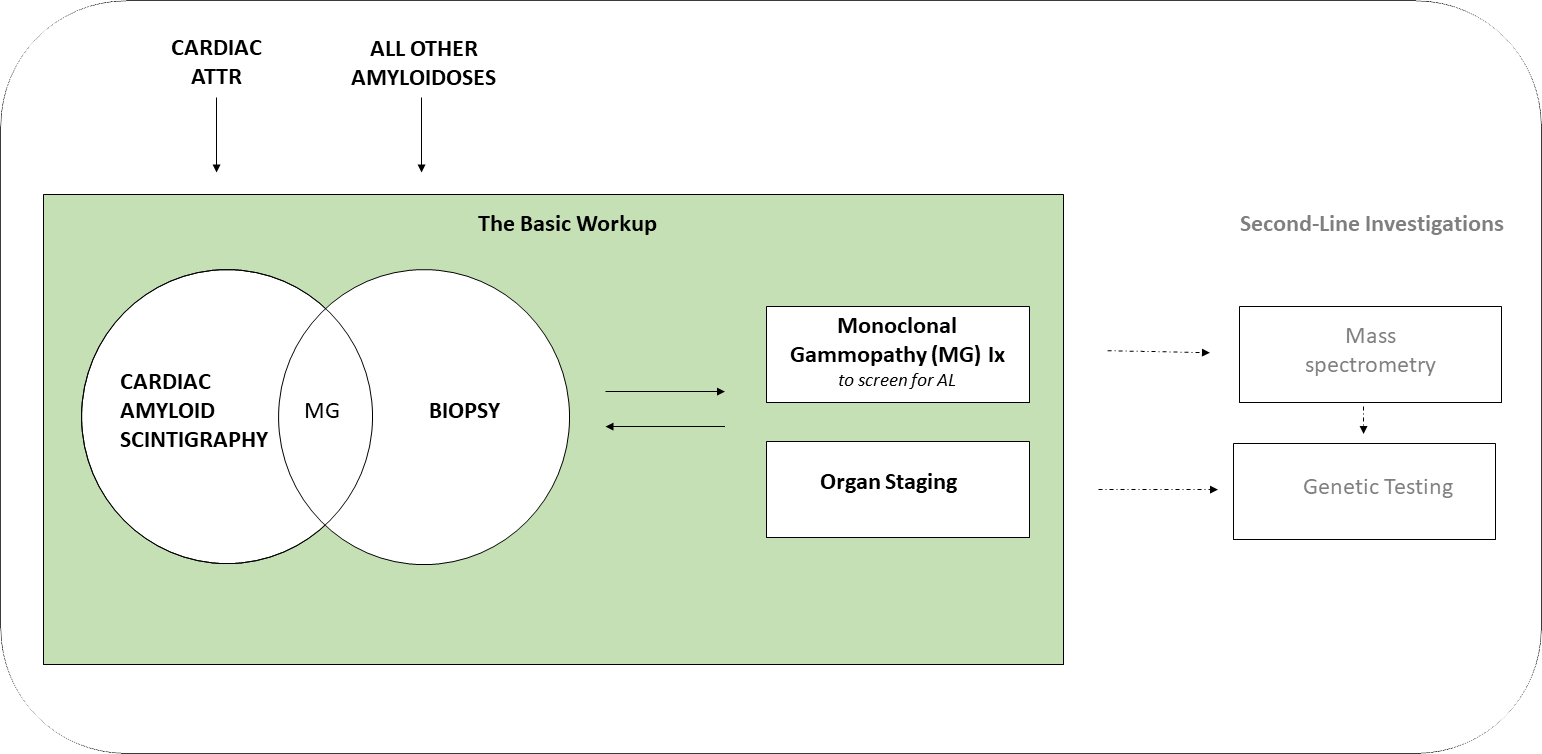
This part of the website discusses the basic workup and management approach of suspected systemic amyloidosis.
The presence of amyloid can now be identified using either;
- Cardiac amyloid bone scintigraphy (for cardiac ATTR)
- OR
- Biopsy (for all other amyloidoses)
All cases still require;
- Monoclonal gammopathy tests (to screen for AL)
- AND
- Organ staging
Some cases may require further amyloid typing testing with;
- Mass spectrometry for protein typing
- Genetic testing for amyloidogenic mutations
The typing of amyloidosis is a clinico-pathologic process requiring multiple correlates to accurately type the amyloidosis. There is no singular test that can definitively type the amyloid in isolation.
The most common and critical pitfalls to be aware of are that;
- Immunohistochemical amyloid typing stains only have a positive predictive value of ≈ 60%
- AND THAT
- Cardiac amyloid bone scintigraphy has a high positive predictive rate for cardiac ATTR amyloidosis but can be positive in cardiac AL and does not exclude non-ATTR cardiac amyloidosis
One of the primary purposes of the State based AAN clinico-pathologic services is to assist with the typing of amyloidosis. Referrals for this purpose are welcomed and strongly encouraged. Performing the basic workup expedites the diagnostic process.
The Basic Workup and Approach: in detail
When to Suspect Amyloidosis
KEY POINT
The question “Could this be Amyloidosis” should be asked when there is;
- Unexplained single organ or multi-organ failure with amyloid-like manifestations
- Presentation with a classical clinical phenotype of one of the amyloidoses
In systemic amyloidosis the vital organs are affected in similar ways no matter the type of amyloidosis;
- Heart: diastolic heart failure with preserved ejection fraction (HFpEF), atrial fibrillation and other dysrhythmias
- Kidneys: proteinuria, nephrotic syndrome, renal failure
- Nerves: length dependent peripheral sensory and motor neuropathy, autonomic neuropathy
- Gastrointestinal tract (GIT): (upper GIT) weight loss, early satiety, peptic ulceration and (lower GIT) diarrhoea, alternating bowel habit, per rectal bleeding, colic
- Liver: hepatomegaly, obstructive liver function test abnormalities
- Spleen: hyposplenism, splenomegaly
The diagnosis of amyloidosis is usually delayed as these organ failure syndromes are commonly caused by other disorders. The key is to consider amyloidosis when there is unexplained;
- Amyloid-like cardiac failure e.g. HFpEF without a history of hypertension (a common presentation of cardiac ATTRwt)
- or
- Multiple amyloid-like organ failure syndromes occurring concurrently e.g. HFpEF and proteinuria without obvious causes (a common presentation of AL)
There are classical “phenotypes” for the systemic amyloidoses that can aid in their identification. Phenotypic presentations for the most common types of amyloidosis are summarised in Table 1. As there is overlap between these phenotypes, they cannot be used to robustly type the amyloidosis.
Table 1: Classical Clinical Phenotypes for Amyloidosis Types or Groups1
| AMYLOID TYPE/GROUP | Fibril Protein | % AAN referrals |
CLASSICAL CLINICAL PHENOTYPE | |
COMMON |
||||
| Wild type transthyretin | ATTRwt | 45 | Male of older age with slowly progressive cardiac infiltration/thickening on TTE presenting with diastolic heart failure (with preserved EF) and/or atrial fibrillation. Notably, ATTRwt does not deposit in the kidneys to cause proteinuria.
Background of (often bilateral) carpel tunnel syndrome. |
|
| Systemic light chain | AL | 30 | Male or female with heart failure and/or nephrotic syndrome without a clear cause.
Commonly ≥ 2 vital organs are involved but single organ involvement is not uncommon. |
|
| Localised amyloid group | AL |
12 |
10 |
Localised AL is most commonly identified within mucosal tissues and presents with site-specific symptoms e.g. intermittent haematuria (bladder), hoarse voice (larynx), rash (skin). |
| ASem1 |
1 |
Semenogelin amyloid is produced in the seminal vesicles and most commonly presents with intermittent haematospermia but is also incidentally detected in prostate tissue. | ||
| AIns |
1 |
Amyloidoma at insulin injection sites from bovine or porcine derived insulin preparations. | ||
UNCOMMON
|
||||
| Hereditary TTR | ATTRv | 5-7 | Mixed heart and nerve disease with one organ predominating.
Nerve disease commonly involves peripheral sensory and motor nerves and autonomic nerves. Endemic in the Portuguese, Swedish, Japanese and Irish but not infrequently occurs “sporadically” in other ethnic groups. |
|
| Serum Amyloid A | AA | 3 | Nephrotic syndrome on a background of an active chronic auto-inflammatory disorder and/or infection. | |
RARE
|
||||
| Leucocyte Chemotactic Factor-2 | ALECT2 | ≈1 | Typically presents with a slowly progressive subnephrotic proteinuria in non-Anglo-Saxon ethnic groups. | |
| Hereditary non-TTR amyloidosis group | AFib
ALys AApoAII AApoAI AGel |
≈1 | The non ATTR hereditary amyloidoses are rare with differing organ tropism patterns depending on the genetic mutation involved.
The most common is AFib which targets the kidneys and presents with nephrotic syndrome and renal failure. |
|
| Immunoglobulin heavy chain | AH | <1 | AH presents similarly to AL. | |
| Aβ2M | β2-Microglobulin wild type | <1 | Joint pain or arthropathy in a heamodialysis patient. This type does not involve the heart or kidneys. | |
Key: A=acquired, ANS= autonomic nervous system, CNS= central nervous system, H= hereditary, HT= hypertension, IHD= ischaemic heart disease, L= localised, PNS= peripheral nervous system, Rare = <1% AAN clinic referrals, S=systemic, TTE= transthoracic echocardiogram, Uncommon = <10% of AAN clinic referrals
Amyloid Identification
KEY POINT
- Cardiac Amyloid Bone Scintigraphy can sensitively and specifically identify cardiac ATTR without the need for biopsy
- A biopsy is required to identify all other types of systemic amyloidosis
- A biopsy is also required for those with both positive uptake on cardiac amyloid scintigraphy and a monoclonal gammopathy
AL and Organ Screening
KEY POINT
- For all amyloidosis cases it remains necessary to perform;
- Monoclonal gammopathy testing to screen for AL
- Organ staging
- These screening procedures are also useful to perform in cases of suspected amyloidosis
Second-Line Investigations
KEY POINT
- Mass spectrometry analysis of the constituent amyloid protein is required when the type of amyloidosis remains uncertain after the basic work-up has been performed
- Genetic screening is recommended when the constituent amyloid protein can be formed by a hereditary process
- Genetic testing is commonly indicated in cardiac ATTR to differentiate between wild type and hereditary ATTR
The Basic Management Approach
KEY POINT
- Management of amyloidosis is divided into supportive care and disease modifying therapy
- Disease modifying therapies can be subdivided into;
- Reducers or suppressors of amyloid protein production
- Targeted molecular therapies
- Amyloid eliminators
- Notably, de-novo or endogenous macrophage clearance of amyloid can also occur
Management of the amyloidoses is divided into disease modifying and supportive therapies. Supportive care is described within “The Organ” sections of this website. Disease modifying therapy can be subcategorised into the following approaches;
- Reducers or suppressors of amyloid precursor protein production. Examples of this approach are;
- Silencing RNA therapy to decrease liver TTR production
- Anti- plasma cell chemotherapy to decrease monoclonal amyloidogenic light chain production in AL
- Disease modifying drugs for Rheumatoid Arthritis driven AA
- Surgical resection of the cells producing localised AL in mucosal tissue
- Targeted molecular therapies directed against the kinetics of amyloid formation
- The best examples of this approach are in ATTR therapy. TTR tetramer dissociation into amyloidogenic monomers is the first step in TTR amyloid fibril formation. Tafamidis and AG110 “stabilise” the TTR tetramer preventing dissociation into amyloidogenic TTR monomers (view the Management tab on this page).
- Amyloid eliminators
- A limited number of therapies have been shown to eliminate amyloid with variable success in early phase research
- “CPHPC” followed by anti-SAP monoclonal antibody holds the most promise as a pan-amyloid eliminator but progress has been stalled in early phase clinical trials.17 This therapy targets a ubiquitous constituent protein in all amyloid deposits (SAP) and triggers macrophage driven clearance.
Amyloid can also be cleared slowly by endogenous de-novo macrophage processes;
- For organ recovery, the rate of production of amyloid needs to be less than the native macrophage clearance rate
- The efficacy of macrophage mediated amyloid clearance varies between individuals
- The likelihood of amyloid clearance also differs between organs
- The macrophage clearance rate is slow and signs of organ recovery can take up to ≈ 18 months
Common Misconceptions
KEY POINT
- Amyloidoses is commonly misdiagnosed
- The most common erroneous assumptions are that;
- Immunohistochemical typing stains have a high degree of accuracy
- Cardiac amyloid bone scintigraphy uptake is confined to transthyretin type cardiac amyloidosis
Amyloidosis is commonly misdiagnosed and one study showed that the misdiagnosis rates for AL was 39%, ATTRwt 26% and ATTRv 29% 18. Common misconceptions regarding the workup of amyloidosis are addressed in this section.
Medical History
Misconception
“There is no family history and so my patient cannot have hereditary amyloidosis”Explanation
Approximately 50% of proband cases of hereditary amyloidosis do not have a clear family history of inherited neuropathy, cardiac failure or sudden cardiac death. 19
Misconception
“There is no inflammatory disorder and so my patient does not have AA amyloidosis”Explanation
Approximately 20-30% AA patients do not have an auto-inflammatory condition aor chronic infection. In most of these cases have obesity appears to be the inflammatory risk factor. 20
Misconception
“My patient with cardiac amyloidosis does not look too sick and has a good exercise tolerance ….so they cannot have cardiac AL.”Explanation
Some cardiac AL patients may have a period of asymptomatic cardiac disease before the amyloid cardiac load becomes clinically significant. Symptom burden is not specific enough to differentiate between cardiac ATTR and cardiac AL.
Misconception
“My patient has had findings of cardiac amyloidosis for several years and so they cannot have AL amyloidosis and they must have cardiac ATTRwt”Explanation
The median time to diagnosis from symptom onset of AL is two years. 21 The pace of disease is not specific enough to differentiate between the amyloidoses.
Anatomical Pathology Identification and Typing
Misconception
“Congo Red Staining was positive/negative report for amyloid on the biopsy and so it is or is/is not present in my patient.”Explanation
There is a low false positive and false negative rate for diagnosis of amyloid by Congo red staining. False positives can occur when a diagnosis of amyloid is based solely on salmon pink staining of extracellular tissues (a not uncommon finding) without confirmation of apple green birefringence under polarised light. False negatives can occur when the Congo Red stain is not properly maintained.
Misconception
“The immunohistochemical stain of my patient’s heart biopsy is positive for AA and so my patient has cardiac AA.”Explanation
Immunohistochemical typing stains only have ≈ 60% positive predictive value. A clearly positive typing result is where there is strong positivity for only one amyloid typing stain and where corollary findings (such as the clinical phenotype, monoclonal gammopathy results and cardiac scintigraphy) support the typing. No one test result can ever be used in isolation to type amyloidosis.
Misconception
“Every amyloid patient’s biopsy needs protein typing by light dissection and mass spectrometry analysis”.Explanation
Mass spectrometry typing of amyloid deposits is indicated in unclear systemic cases where the immunohistochemical typing stains are non-contributory and/or do not match the other corollary findings. Mass spectrometry is usually not required in localised amyloid and in a significant proportion of cases of systemic AL.
Cardiac Amyloid Bone Scintigraphy
Misconception
“My patient has a positive cardiac amyloid bone scintigraphy scan and so they must have cardiac ATTR”.Explanation
Some AL patients may also show uptake on cardiac amyloid scintigraphy. Every patient who has a positive cardiac amyloid scintigraphy scan must therefore undergo monoclonal gammopathy testing to screen for the presence of AL. If there is a monoclonal gammopathy then a biopsy is required to distinguish between 1. ATTR with an intercurrent uninvolved MGUS vs 2. Cardiac AL.
Organ staging
Misconception
“I need to biopsy every organ to confirm the presence of amyloid involvement.”Explanation
Once amyloid is detected at one site, organ staging can be performed using non-invasive techniques.
Misconception
“Every patient with suspected cardiac amyloidosis needs a cardiac MRI”Explanation
In most cases, sufficient features of cardiac amyloidosis can be identified by the combination of clinical assessment, cardiac biomarkers, ECG and TTE (with global longitudinal strain) to support proceeding to diagnostic tests. Cardiac MRI is a useful adjunct when other potential disorders such as hypertension and non-amyloid cardiomyopathies are being considered. Cardiac MRI cannot confirm the presence of cardiac amyloid nor type cardiac amyloid. For these purposes, cardiac amyloid bone scintigraphy (for cardiac ATTR) or biopsy (for all other types of cardiac amyloidosis) are required.
Monoclonal Gammopathy Tests
Misconception
“My cardiac amyloid patient has a monoclonal gammopathy and so they have cardiac AL”.Misconception
“My patient only has a mildly elevated (kappa or lambda) light chain and so they cannot have AL”.Explanation
The degree of light chain elevation is lower in AL than in light chain multiple myeloma. The range of the monoclonal free light chain level at diagnosis for patients with systemic AL is between 30-500mg/L. 22 Approximately 13-19% of AL patients will have a low amyloidogenic free light chain level at presentation. (“Low level” is defined as a <50mg/L difference between the involved free light chain and uninvolved free light chain). 23
Misconception
“My patient has a slightly elevated kappa light chain and so they have AL amyloidosis”.Explanation
SFLC results can be affected by both laboratory clinical issues. Clinically, kappa and lambda free light chains can be elevated by renal impairment and inflammation. Renal impairment leads to a disproportionately greater increase in physiologic kappa free light chain as monomeric kappa free lights are renally excreted quicker than dimeric lambda free light chains. In end-stage kidney disease, kappa: lambda FLC ratios of up to 3.1 have been reported. 24
Genetic screening
Misconception
“Every patient with amyloidosis needs genetic screening”.Explanation
Many types of amyloidosis are not inherited. Systemic AL and AA are acquired amyloidoses where genetic testing is not required. There are now improved amyloid typing techniques along with better knowledge of which amyloidosis types may be hereditary to indicate when genetic testing is required.
Misconception
“ATTR patients do not require genetic screening as inherited ATTRv is rare and there is “nothing” we can do about it”.Explanation
A significant minority of ATTR cases carry an amyloidogenic TTR gene mutation. In one AAN clinic, ≈10% of the ATTR cohort and ≈ 5% of the cardiac ATTR cohort carried a TTR gene mutation. 25 In the UK, 17% of screened ATTR cases had an inherited TTR mutation. 26 Disease modifying therapies are now becoming increasingly available for those with ATTR and their kindred.